
Abstract
Background and Objectives
OTX015 (MK-8628) is a novel inhibitor of the bromodomain and extraterminal (BET)-bromodomain (BRD) protein family, binding specifically to bromodomains BRD2/3/4 and impacting the epigenetic regulation of several oncogenes. We characterized the pharmacokinetics of this first-in-class BET-BRD inhibitor administered as a single agent, including population pharmacokinetic modelling.
Methods
A dose-escalation, phase Ib study was performed with oral OTX015 in patients with haematologic malignancies, at doses starting from 10 mg once daily (QD) with continuous or discontinuous schedules. Five or eight blood samples were collected per patient for pharmacokinetic analysis. OTX015 plasma concentrations were determined using validated ultra-performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) and analysed using a nonlinear mixed-effects modelling software program. A population pharmacokinetic model was fitted to the data, and patient demographics and clinical chemistry parameters were tested as predictive covariates on the model parameters.
Results
Blood samples were analysed from 81 patients treated with OTX015 at doses ranging from 10 to 160 mg QD or 40 mg twice daily (BID), and 633 time–plasma concentrations were available for analysis. A one-compartment open model with linear elimination adequately described OTX015 pharmacokinetics. The most significant covariate was lean body mass (LBM), which decreased the between-subject variability in apparent total body clearance (CL) and the volume of distribution (V). The estimated pharmacokinetic parameters were the absorption rate constant (k a) = 0.731 h−1, V = 71.4 L and CL = 8.47 L·h−1.
Conclusion
The pharmacokinetics of oral OTX015 in patients with haematologic malignancies can be described with a one-compartment model. Population pharmacokinetic modelling of OTX015 plasma concentrations showed that LBM influences V and CL. These findings do not suggest the need for dose adjustment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This is the first clinical description of the pharmacokinetics, including population modelling, of the oral bromodomain inhibitor OTX015/MK-8628. |
The best pharmacokinetic model was a one-compartment model. Lean body mass was the only covariate significantly affecting OTX015/MK-8628 pharmacokinetics (the volume of distribution and apparent total body clearance), but it did not have a clinically relevant impact (no dosing adjustment is required). |
1 Introduction
Bromodomain and extraterminal (BET)-bromodomain (BRD) proteins, such as BRD2/3/4 and BRDT (BRD testis-specific protein), bind to acetylated lysines on histones. This contributes to the assembly of transcriptional co-regulators on target gene promoters and enhancers, regulating the expression of key oncogenes, such as c-MYC, CDK6, BCL2 and IL7R [1]. A novel class of epigenetic compounds selectively targeting BET-BRD proteins in haematologic malignancies and solid tumour models represents a new opportunity in oncology drug development [2–5]. OTX015 (MK-8628), 2-[(6S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f]-[1, 2, 4] triazolo [4,3-a] [1, 4] diazepin-6-yl]-N-(4-hydroxyphenyl)-acetamide dehydrate (Fig. 1) selectively binds to BRD2/3/4 proteins, hindering transcription of associated oncogenes [6], as has been extensively reported for its derived-analogue JQ1 [7]. OTX015 inhibits BRD proteins in several preclinical models [6] and shows significant in vitro and in vivo antitumour activity in a number of haematologic malignancy models [6], including leukaemia [8], multiple myeloma [9] and lymphomas [10], as well as in paediatric and solid tumour models. The pharmacokinetics of OTX015 in these preclinical models exhibited good bioavailability and linear pharmacokinetics.

Chemical structure of OTX015
An understanding of the biological fate of drugs is a cornerstone of their clinical development [11, 12]. To date, characterization of the pharmacokinetic profile of OTX015 in patients has not been investigated. OTX015 is currently under investigation in three phase I trials in adult patients, the first of which is a phase 1b dose-escalation study in patients with haematologic malignancies (ClinicalTrials.gov study ID NCT01713582). The objectives of this multicentre phase Ib dose-escalation study were to assess the safety, tolerability and pharmacokinetic profile of OTX015 as an oral single agent. We used a population pharmacokinetic approach, using data from all haematologic malignancy patients simultaneously to describe the pharmacokinetics of OTX015 and to determine inter-individual and intra-individual variability in OTX015 pharmacokinetic parameters to guide OTX015 dosing in cancer patients, in support of the clinical development of this first-in-class BET-BRD inhibitor.
2 Methods
2.1 Ethics
The study was approved by national ethics committees in all countries where the study was performed, and it was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation (ICH) guidelines for Good Clinical Practice [13]. Written informed consent was obtained from all patients before any study procedures were performed. The study is registered at ClinicalTrials.gov (study ID NCT01713582).
2.2 Study Population
Patients were included in nine centres worldwide between January 2013 and September 2014 in the dose-escalation part of a phase Ib open-label study with a cohort-expansion phase (the latter part of the study is not included in this evaluation). Two dose-escalation cohorts composed of patients with acute leukaemia (AL) or other haematologic malignancies (OHM) were assessed independently and in parallel. The main eligibility criteria were a diagnosis of AL or OHM in patients aged 18 years or older with failure under all standard therapies or for whom standard treatment was contraindicated. Patients had to have a Eastern Cooperative Oncology Group (ECOG) performance status of 0–2 and adequate liver, renal and bone marrow function (the latter was applicable in OHM patients only).
2.3 Dosing
OTX015 was supplied as gelatine capsules (10 or 20 mg) containing OTX015 free base as a solid molecular dispersion, and was administered orally under fasted conditions. For each patient cohort (AL and OHM), dose-escalation was performed using a conventional 3 + 3 design with a modified Fibonacci dose escalation, at a starting dose of 10 mg once daily (QD) up to 160 mg QD (Table 1). A 40 mg twice-daily (BID) dose was also evaluated. For AL patients, OTX015 was taken over 14 consecutive days followed by a 7-day rest period, while for OHM patients, it was taken continuously without a planned rest period (Fig. 2). Schedule optimization was performed at selected dose levels.

Treatment schedule for both cohorts [acute leukaemia (AL) and other haematological malignancy (OHM) patients]. Days of treatment are shaded in blue, and the 7-day rest period is unshaded. PK pharmacokinetic
2.4 Sampling Schedule
Venous blood samples (3 mL) were collected according to two sampling schedules with the QD administration, complete and limited. The complete schedule consisted of samples collected at time 0 (predose) and at 1, 4, 8, 12 and 24 h after the first OTX015 administration, plus at either 10 or 16 h on days 1 and 2 in the first three patients at each dose level. The limited schedule consisted of samples collected at time 0 (predose) and at 1, 4, 6 and 8 h on day 1 for other patients treated at this dose level. For all patients treated with OTX015 at 40 mg BID, samples were collected at time 0 (predose) and at 20 min, 1 h, 2 h 15 min, 3 h 15 min, 9 h, 12 h and 24 h. In all patients, trough concentrations were assessed immediately before drug intake on days 8, 15 and 22 (Fig. 2). Blood samples were collected in ethylenediaminetetraacetic acid (EDTA) tubes, and plasma aliquots were stored at −30 °C until analysis in a centralized laboratory.
2.5 Analytical Methods
OTX015 concentrations were determined using a validated Acquity Ultra Performance LC system (Waters, Milford, MA, USA) coupled with a tandem mass spectrometry detection method, as described previously [14]. Reversed-phase chromatographic separation was performed on a BEH C18 ultra-performance liquid chromatograph (UPLC) 1.7 µm, with a 2.1 × 50 mm column. The mobile phases were water with 5 mM ammonium acetate and acetonitrile. The gradient elution was performed at a flow rate of 0.5 mL·min−1. The lower limit of quantification of OTX015 was 1 ng·mL−1, with 10.80 % precision and 94.67 % accuracy.
2.6 Pharmacokinetic Modelling
Data were analysed using the nonlinear mixed-effects modelling software program Monolix version 4.3.2s [15]. Parameters were estimated by computing the maximum likelihood estimator without any approximation of the model (no linearization), using the stochastic approximation expectation maximization (SAEM) algorithm combined with a Markov chain Monte Carlo (MCMC) procedure. The number of MCMC chains was fixed at a value of 10 for all estimations of OTX015. Between-subject variability (BSV) was ascribed to an exponential model, and different error models were tested to describe the residual variability. All values calculated below 1 ng·mL−1 were considered to be below the lower limit of quantification (BLQ) in the analysis and were censored. Outlier data were identified on an initial visual inspection of the raw data and were not included in the analysis.
Ten covariates were investigated: gender and cohort (considered as categorical covariates), and age, height, weight, aspartate aminotransferase (AST), alanine aminotransferase (ALT), albumin, ideal body weight (IBW) and lean body mass (LBM) (considered as continuous covariates). All subjects had complete covariate information (Table 2).
For model development, the effects of covariates were assessed individually and simultaneously on the pharmacokinetic parameters. All combinations of covariates were guided and the best model was chosen according to standard goodness-of-fit plots [observed concentrations versus population- and individual-predicted concentrations, individual and population-weighted residuals, and a visual predictive check (VPC)], precision of estimations, the Bayesian information criterion (BIC) and the Akaike information criterion (AIC). The VPC was performed to ensure that the simulations from the model reproduced the observed data. It was based on 1000 simulations for the 5th and 95th percentiles with a 90 % confidence interval.
2.7 Pharmacokinetic–Pharmacodynamic Modelling
Pharmacokinetic–pharmacodynamic (PK-PD) modelling was performed using the model published by Friberg et al. [16] in order to relate OTX015 pharmacokinetic parameters to a pharmacodynamic effect, thrombocytopenia. This PK-PD model, based on a semi-mechanistic structural model, consisted of one compartment representing proliferative cells (PROL), three transit compartments with maturing cells (transit) and a compartment of observed circulating platelets. This model mimics the maturation chain of progenitor cells in bone marrow to differentiated thrombocytes in the systemic circulation. Transit compartments with a rate constant (k tr) stood for the time delay between administration and the observed effect. The drug effect was defined as a maximum effect (E max) model [E max × Conc/(IC50 + Conc), where Conc is the concentration and IC50 is the concentration of the drug producing 50 % inhibition] on the proliferation rate constant (k prol) of the progenitor cells. E max was fixed at a value of 1. A feedback loop, defined as the ratio between the baseline OTX015 concentration and the concentration at any time raised to the power ‘gg’, was used to describe the rebound of cells [16]. Analyses were performed using platelet counts available in the OHM pharmacokinetic population.
3 Results
3.1 Patient Characteristics for Pharmacokinetic Modelling
Between January 2013 and September 2014, 86 patients were treated over six OTX015 dose levels (10, 20, 40, 80, 120 and 160 mg QD, and 40 mg BID). Among the 86 patients treated in the dose escalation, 81 (40 AL patients and 41 OHM patients) were evaluable for pharmacokinetics; 52 were male and 29 were female, with a median weight of 70 kg (range 44–127), as shown in Table 2. A total of 633 plasma concentrations were available for analysis, 24 of which were BLQ.
3.2 OTX015 Pharmacokinetic Parameters
The mean individual pharmacokinetic parameters obtained after single dosing on day 1 derived from empirical Bayesian estimates (EBE) are summarized in Table 3 for each dose level. The median elimination half-life (t ½) was approximately 5.7 h after single-dose administration. Increases in the maximum plasma drug concentration (C max), area under the plasma concentration–time curve (AUC) from 0 to 24 h (AUC0–24 h) and predose drug concentration (C min) were generally dose proportional with single doses up to 120 mg (Table 3).
3.3 OTX015 Population Pharmacokinetic and Pharmacodynamic Modelling
On the basis of this visual inspection, along with an estimation of the pharmacokinetic parameters, a one-compartment model was considered to best describe OTX015 plasma concentrations. The model was composed of three parameters: the absorption rate constant (k a), volume of distribution (V) and apparent total body clearance (CL). As no intravenous data were available, the initial bioavailability (F) was set at 1, meaning that CL and V were interpreted as the apparent total body clearance (CL/F) and apparent volume of distribution (V/F), respectively. A two-compartment model was also considered but showed a poor estimation of the pharmacokinetic parameters [relative standard error (RSE) >50 %]. Between-subject variability values (BSV and η) were estimated for all pharmacokinetic parameters. Residual variability was described by a constant error model because a proportional or combined error model did not improve the estimation or goodness-of-fit plots (data not shown). For the base model, the k a, V and CL estimates were 0.66 h−1, 56.1 L and 7.54 L·h−1 (RSEs 13, 6 and 6 %, respectively) and the corresponding BSVs [standard deviations (SDs)] were 0.89, 0.38 and 0.41 (RSEs 12, 14 and 11 %, respectively).
The main covariate effect related to body size descriptors. Given that the patients treated in the phase Ib study had a wide range of body mass indices, multiple body size descriptors were investigated: body weight (BW), IBW (see Eq. 1 [17]) and LBM (see Eq. 2 [17]).
where x = 105 for women and x = 100 for men.
where y = 1.07 and z = 148 for women, and y = 1.1 and z = 128 for men.
Table 4 summarizes the model-building steps. LBM was found to be the best size descriptor when CL and V were normalized for LBM to a 60 kg patient according to an allometric scaling rule. The impact of LBM on OTX015 pharmacokinetics was further assessed by classifying patients into three categories based on their quantile (<25th, 25th−75th and >75th). OTX015 exposure was also simulated using Bayesian post hoc pharmacokinetic parameters. The mean (minimum–maximum) values of the OTX015 AUC, taking into account LBM, were: 17,700 (1005−41,540), 17,780 (1060−91,620) and 17,390 (724−37,050) ng·h·mL−1 for the <25th, 25th−75th and >75th quantiles, respectively.
A significance correlation in individual estimates of CL and V was also observed. This reduced the global BSV and increased the goodness-of-fit criteria. A decrease in AIC/BIC values demonstrated that model 4 was the best descriptor of OTX015 pharmacokinetics, reflecting the impact of LBM. No significant influence was observed for the other covariates that were investigated, including gender and patient cohort.
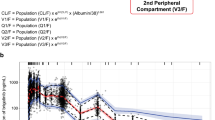
The improvement of fit from a covariate-free model to the final model is illustrated in Fig. 3a and b. The final covariate model for clearance was CL i = 8.47 × (LBM i /60)0.75, where 8.47 denotes the population typical value (TV) of CL and corresponds to the CL of a patient with an LBM of 60 kg. Figure 3c shows the results of the VPC for the final OTX015 population model at the 80 mg QD dose level.

Diagnostic plots of observed (obs) OTX015 concentrations, expressed in ng·mL−1, versus the model population predictions (pop pred left) and individual predictions (ind pred right) for the covariate-free model (a) and the final model (b). The blue dots correspond to observed values, the pink line is the line of unity and the light blue line is the spline
The final model was validated according to the AIC and BIC, as well as the BSV decrease. VPCs based on 1000 simulations showed the good predictive performance of the final model (Fig. 4). A conditional weighted residual plot was used to evaluate the final population pharmacokinetic model (Fig. 5). Considering that the model predictions versus observations were symmetrically and evenly distributed around the line of identity, we concluded that the model adequately describes OTX015 population pharmacokinetics. In the final model, the k a, V/F and CL/F estimates were 0.731 h−1, 71.4 L and 8.47 L·h−1, respectively (RSEs 12, 6 and 5 %, respectively) and the corresponding BSVs were 0.77, 0.36 and 0.38, respectively (RSEs 12, 13 and 11 %, respectively) (Table 5).

Visual predictive check for the final plasma OTX015 population model at 80 mg once daily from 0 to 24 h. The upper and lower lines represent the 90 % confidence interval for 1000 simulated samples, and the blue dots correspond to observed values. emp empirical, out outlier, PI prediction interval, prctile percentile

Individual weighted residuals (IWRES) and normalized prediction distribution error (NPDE) versus time (top row) and versus predicted (pred) concentration (middle row), and empirical distributions of the weighted residuals with the standard Gaussian probability density function (pdf bottom row). cond conditional
PK-PD modelling was performed in terms of thrombocytopenia, a toxicity that was considered to be dose limiting in the OHM population, since AL patients have low platelet counts as a function of their disease. Out of 41 OHM patients, 37 were included in the pharmacodynamic modelling, with 424 platelet counts available for analysis. The pharmacodynamic parameters were well estimated, showing good precision (Table 6).
4 Discussion
We report here for the first time the characterization of the pharmacokinetic profile, including population modelling, of the oral small-molecule BET-BRD inhibitor OTX015 administered in a first-in-man phase Ib study in patients with haematologic malignancies. A preliminary pharmacokinetic analysis performed after 42 patients (from both AL and OHM cohorts combined) had been treated at dose levels of 10, 20, 40 and 80 mg QD and 40 mg BID [18] suggested that the OTX015 plasma concentration–time curves were well described by a one-compartment open model with linear elimination. The present final pharmacokinetic analysis performed in all 81 patients treated in the dose-escalation part of the study and evaluable for pharmacokinetic analysis revealed that LBM is statistically superior to BW in explaining OTX015 pharmacokinetic variability, while albumin did not show a significant or relevant influence on the pharmacokinetics in the current analysis, as initially suspected from the preliminary analysis. The final covariate model that considered LBM reduced the inter-individual variability of OTX015 pharmacokinetic parameters in comparison with the base model. These results indicate that the covariate model accurately describes the relationship between individual pharmacokinetic parameter estimates and patient baseline LBM. Nonetheless, while LBM was identified as statistically significant in the population pharmacokinetic analysis, the impact on OTX015 CL across the evaluated dose range was low. On the basis of the population pharmacokinetic covariate analysis, comparison of the expected OTX015 exposure simulated from the Bayesian post hoc pharmacokinetic parameters and a low BSV decrease suggests that further dose adjustment based on LBM is unlikely to have a clinically meaningful impact on OTX015 exposure. The lipophilic nature of the chemical structure of OTX015 (Fig. 1) may explain the large volume of distribution of OTX015, which, in turn, suggests widespread tissue diffusion. The k a value indicates rapid absorption of OTX015, which is consistent with the time to reach C max (t max) value (between 1 and 4 h).
In the final predictive model, the k a, V/F and CL/F estimates were 0.731 h−1, 71.4 L and 8.47 L·h−1, respectively (RSEs 12, 6 and 5 %, respectively) and the corresponding BSVs were 0.77, 0.36 and 0.38, respectively (RSEs 12, 13 and 11 %, respectively). The relatively slow elimination of OTX015 with a mean t½ of 5.7 hours suggests that steady state would be reached after 2 days of dosing.
The safety and tolerability profile of OTX015 in haematologic malignancy patients in this phase Ib study was reported as manageable [19]. Thrombocytopenia was considered to be the principal dose-limiting toxicity in OHM patients. The estimated IC50 value obtained with the Friberg PK-PD model was relatively high (1800 ng·mL−1), indicating that haematotoxicity could be observed at high OTX015 dose levels. Other pharmacodynamic parameters obtained with this model were consistent with those published for a sequential model used to predict thrombocytopenia with a histone deacetylase (HDAC) inhibitor in both solid tumour patients and lymphoma patients [20]. Patients are currently being treated in the expansion cohort part of this ongoing phase Ib study, and the safety, efficacy and an extended analysis of the pharmacokinetic parameters will be reported separately. Additional clinical studies have been initiated in patients with selected solid tumours (ClinicalTrials.gov study ID NCT02259114) and glioblastoma multiforme (ClinicalTrials.gov study ID NCT02296476). Further characterization of the pharmacokinetic profile of single-agent OTX015 in these populations will also be performed.
5 Conclusion
This pharmacokinetic analysis confirms the oral availability of OTX015 when it is administered as a free-base solid molecular dispersion formulation in patients with haematologic malignancies. We describe here the first population pharmacokinetic model of this novel BET-BRD inhibitor. A one-compartment model with linear elimination adequately describes the pharmacokinetics of OTX015. Population pharmacokinetic modelling allowed estimation of the mean population pharmacokinetic parameters of OTX015, showing that LBM influences the V and CL of OTX015. However, OTX015 dose adjustment on the basis of LBM is not necessary. Treatment of patients with selected haematologic malignancies in expansion cohorts at the dose of 80 mg QD, 14 days on/7 days off, is ongoing, and the proposed population pharmacokinetic and pharmacodynamic models will be further evaluated in patients from this and future clinical studies.
References
Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:2843–52.
Zhang G, Liu R, Zhong Y, Plotnikov AN, Zhang W, Zeng L, et al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem. 2012;287:28840–51.
Herrmann H, Blatt K, Shi J, Gleixner KV, Cerny-Reiterer S, Müllauer L, et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia (AML). Oncotarget. 2012;1588–99.
Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res. 2013;19:1748–59.
Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, et al. Efficacy of BET bromodomain inhibition in KRAS-mutant non-small cell lung cancer. Clin Cancer Res. 2013;19:6183–92.
Noel JK, Iwata K, Ooike S, Sugahara K, Nakamura H, Daibata M. Development of the BET bromodomain inhibitor OTX015 (abstract no. C244). Mol Cancer Ther. 2013;12:C244.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–73.
Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci. 2011;108:16669–74.
Todaro M, Boi M, Vurchio V, Ercole E, Machiorlatti R, Messana K, et al. OTX015, a novel BET inhibitor, is a promising anticancer agent for multiple myeloma (abstract no. 5531). Cancer Res. 2014;74:5531.
Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, et al. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin Cancer Res. 2015;21:1628–38.
Wagner JG. History of pharmacokinetics. Pharmacol Ther. 1981;12:537–62.
Vozeh S, Steimer J-L, Rowland M, Morselli P, Mentré F, Balant L, et al. The use of population pharmacokinetics in drug development. Clin Pharmacokinet. 1996;30:81–93.
Althoff K, Beckers A, Bell E, Nortmeyer M, Thor T, Sprussel A, et al. A Cre-conditional MYCN-driven neuroblastoma mouse model as an improved tool for preclinical studies. Oncogene. 2015;34:3357–68.
Odore E, Lokiec F, Weill S, Noel JK, Herait P, Bekradda M, et al. Development and validation of an UPLC–MS/MS method for quantitative analysis of OTX015 in human plasma samples. Anal Methods. 2014;6:9108–15.
Kuhn E, Lavielle M. Maximum likelihood estimation in nonlinear mixed effects models. Comput Stat Data Anal. 2005;49:1020–38.
Friberg LE, Henningsson A, Maas H, Nguyen L, Karlsson MO. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol. 2002;20:4713–21.
De Baerdemaeker LE, Mortier EP, Struys MM. Pharmacokinetics in obese patients. Contin Educ Anaesth Crit Care Pain. 2004;4:152–5.
Odore E, Rezai K, Riveiro E, Bourdel F, Herait P, Cvitkovic E, et al. A phase I pharmacokinetic study of OTX015 for the treatment of patients with hematologic malignancies (abstract no. LB-231). Cancer Res. 2014;74:LB-231.
Stathis A, Quesnel B, Amorim S, Thieblemont C, Zucca E, Raffoux E, et al. Results of a first-in-man phase I trial assessing OTX015, an orally available BET-bromodomain (BRD) inhibitor, in advanced hematologic malignancies (abstract no. 5LBA). Eur J Cancer. 2014;50:196.
Chalret du Rieu Q, Fouliard S, White-Koning M, Kloos I, Chatelut E, Chenel M. Pharmacokinetic/pharmacodynamic modeling of abexinostat-induced thrombocytopenia across different patient populations: application for the determination of the maximum tolerated doses in both lymphoma and solid tumour patients. Invest New Drugs. 2014;32:985–94.
Financial Support
This study was funded by Oncoethix SA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
EC and PH are founders of Oncoethix. EO, MB, FB, CK and MER are employees of Oncology Therapeutic Development. FL, ER, AS, CT, BQ, DC and KR have no conflicts of interest that are directly relevant to the content of this manuscript.
Additional information
Oncoethix GmbH is a wholly owned subsidiary of Merck Sharp & Dohme Corp. (formerly Oncoethix SA)
Rights and permissions
About this article

Cite this article
Odore, E., Lokiec, F., Cvitkovic, E. et al. Phase I Population Pharmacokinetic Assessment of the Oral Bromodomain Inhibitor OTX015 in Patients with Haematologic Malignancies. Clin Pharmacokinet 55, 397–405 (2016). https://doi.org/10.1007/s40262-015-0327-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0327-6