
Abstract
Background and Objectives
The effect of ontogeny in drug-metabolizing enzymes on pediatric pharmacokinetics is poorly predicted. Voriconazole, a potent antifungal, is cleared predominantly via oxidative metabolism and exhibits vastly different pharmacokinetics between adults and children. A physiologically based pharmacokinetic (PBPK) model was developed integrating hepatic in vitro metabolism data with physiologic parameters to predict pharmacokinetic parameters of voriconazole in adult and pediatric populations.
Methods
Adult and pediatric PBPK models integrated voriconazole physicochemical properties with hepatic in vitro data into the models. Simulated populations contained 100 patients (10 trials with 10 patients each). Trial design and dosing was based on published clinical trials. Simulations yielded pharmacokinetic parameters that were compared against published values and visual predictive checks were employed to validate models.
Results
All adult models and the pediatric intravenous model predicted pharmacokinetic parameters that corresponded with observed values within a 20 % prediction error, whereas the pediatric oral model predicted an oral bioavailability twofold higher than observed ranges. After incorporating intestinal first-pass metabolism into the model, the prediction of oral bioavailability improved substantially, suggesting that voriconazole is subject to intestinal first-pass metabolism in children, but not in adults.
Conclusions
The PBPK approach used in this study suggests a mechanistic reason for differences in bioavailability between adults and children. If verified, this would be the first example of differential first-pass metabolism in children and adults.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Previous clinical trials have shown marked differences in voriconazole pharmacokinetic parameters between adults and children without providing a mechanistic basis for these disparities. |
The simulations using the physiologically based pharmacokinetic (PBPK) models suggest that intestinal first-pass metabolism is an important factor that contributes to lower oral bioavailability of voriconazole in children compared with adults. |
This study provides a new paradigm for predicting therapeutic doses in children, in which adult in vitro metabolism are related to in vivo pharmacokinetics to create and validate a PBPK model and then adapted to use in vitro metabolism by pediatric tissue for predicting pharmacokinetics in children. |
The most important use of this approach is to develop safe and effective pediatric dosing regimens, despite limited clinical data. |
1 Introduction
Voriconazole (Vfend®; Pfizer, New York, NY, USA) is a potent triazole antifungal commonly used in patients with life-threatening invasive fungal infections [1]. Since its approval in 2002, studies demonstrated that voriconazole improves survival and produces fewer severe adverse effects compared with amphotericin B, the original standard of care for invasive aspergillus infections [2], making it first-line treatment for patients of all ages. However, voriconazole displays wide variability and significant pharmacokinetic differences in clearance, exposure, and bioavailability between adults and children, with no consensus on pediatric doses that match adult exposures, thereby limiting successful therapy [3–6].
Voriconazole is extensively metabolized by the liver, with only 2 % of the parent excreted in urine [7]. Oral bioavailability of voriconazole in adults is ~96 %, while pediatric bioavailability is 45–66 % [4, 6, 7]. Voriconazole exhibits threefold higher clearance in pediatric patients compared with adults, and over a narrow dose range of 3–5 mg/kg, it displays non-linear pharmacokinetics in adults but not in children suggesting saturation of metabolic enzymes [6, 7]. Developmental changes in these drug-metabolizing enzymes (DMEs) have been implicated in the pharmacokinetic differences between children (aged 2–10) and adults [8–11]. In vitro studies in our laboratory revealed developmental changes in these DMEs showing that two cytochrome P450 enzymes (CYP), CYP3A4 and CYP2C19, and flavin-containing mono-oxygenase (FMO) 3 contribute to 50, 35 and 15 % of voriconazole metabolism in adults, respectively, vs. 20, 50 and 30 % in children. Hepatic intrinsic clearance (CLint) scaled to in vivo clearance accounted for 80 % of clinically observed values, suggesting that the above differences conferred the increased clearance of voriconazole in children [12].
Although scaling of intrinsic clearance values explains differences in voriconazole clearance between adults and children, it does not provide insights into differences in other pharmacokinetic parameters between these two populations. This study describes a physiologically based pharmacokinetic (PBPK) model for voriconazole that integrates in vitro hepatic metabolism data, voriconazole physicochemical properties, and physiologic parameters associated with adult and pediatric populations. The models were tested against clinically observed data. The model predictions of pharmacokinetics in children led to a hypothesis that intestinal first-pass metabolism of voriconazole in children would explain its lower bioavailability in children compared with adults. PBPK modeling has proven to be useful in understanding mechanisms underlying the differences in pharmacokinetics of voriconazole between adults and children, and can be applied to predict the influence of drug-drug interactions, genetic polymorphisms, and other factors on voriconazole disposition in children.
2 Materials and Methods
2.1 Model Structure
Metabolic assays to generate hepatic in vitro pharmacokinetic parameters, V max (maximum rate of metabolism) and K m (Michaelis–Menten constant), for voriconazole from healthy adult and pediatric tissues were adapted from previously published work [12].
Intial models were created in Berkeley Madonna (version 8.3.18; University of California at Berkeley) to assess linearity. A perfusion-limited model (Supplementary Figure 1) incorporated physicochemical properties (Table 1), with tissue distribution compartments based on published data [13]. Age-dependent physiologic characteristics were averaged between male and female patients [14, 15]. To assess linearity, multiple intravenous infusion doses of 3–6 mg/kg were simulated for both adult and pediatric populations and area under the plasma-concentration curve was calculated using Phoenix WinNonlin (version 6.2, Certara). The proportionality of the models was assessed by dividing the area under the plasma concentration-time curve (AUC) of 4, 5, and 6 mg/kg doses by AUC of the 3 mg/kg dose, and comparing it with the fold change in dose. Finally, clearance and bioavailability were assessed for these simulations.
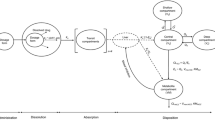
Next, a population-based model was developed employing Simcyp (version 12.1; Certara) using a step-wise approach (Fig. 1). A perfusion-limited model incorporating adult clearance values and physiologic characteristics was created first using adult intravenous infusion dosing of 3, 4, and 5 mg/kg infused over 1 h and dispensed every 12 h for 7 days. Fasting oral dosing was simulated for 200 mg dispensed every 12 h for 7 days. Similarly, a pediatric model was created using in vitro V max and K m generated from pediatric (2–10 years old) liver microsomes and the Simcyp Paediatric Simulator®. Pediatric intravenous infusion simulated doses of 4 and 6 mg/kg infused every 12 h for 7 days at a maximal rate of 3 mg/kg/h and oral dosing of 4, 6, and 8 mg/kg dispensed every 12 h for 4 days. Trial designs and dosing were based on published clinical trials and simulated populations contained 100 patients each (10 trials with 10 patients per trial) [5, 16, 17]. Simulations integrated the age ranges and male-to-female ratios recorded for each respective trial. Physiologic characteristics were predicted applying Method 1 (Poulin and Theil). Method 1 best describes unionized compounds in plasma, such as voriconazole, and predicts compound distribution based on tissue-specific parameters [18, 19]. Physicochemical properties and absorption values, along with in vitro data, were combined into a full PBPK model using whole organ metabolic clearance. In vitro voriconazole metabolic rates generated are listed in Table 1 along with other pertinent parameters employed in model development [4, 7, 12, 20, 21].

Experimental workflow for the bottom-up approach to scaling voriconazole from adults to the pediatric population. IV intravenous, PK pharmacokinetic, PBPK physiologically based pharmacokinetic
2.2 Model Validation
Simulations yielded pharmacokinetic parameters that were compared against published values expressed as geometric means. First, models were validated by comparing the predicted pharmacokinetic parameters against observed pharmacokinetic parameters. Given the immense clinical variability in voriconazole pharmacokinetics in both the adult and pediatric population, differences <50 % were considered acceptable. If the predicted parameters were within 50 % of the observed values, then visual predictive checks were employed to confirm that the model predicted observed concentrations and standard deviation. Concentration-time profiles from a food effect trial conducted in healthy volunteers and a trial in immunocompromised children were digitized using GetGraph data digitizer version 2.25.0.32. Observed voriconazole concentrations were superimposed on predicted mean and 90 % confidence intervals. Last, a distributional analysis of hepatic and intestinal CYP enzymes from simulated trials was generated [22].
2.3 Sensitivity Analysis
Sensitivity analyses, used to assess factors that could influence model performance, were performed for gastric emptying time and small intestinal transit time, both of which could significantly influence pharmacokinetic parameters after enteral absorption. Both values were replicated with a tenfold change in the mean to determine if either parameter significantly affected pertinent pharmacokinetic parameters of AUC and maximum concentration (C max).
3 Results
3.1 Simulations to Assess Linearity
In Berkeley Madonna, non-linear behavior was predicted by the adult model as evidenced by greater than proportional increase in AUC (1.7-, 2.7-, and 4.2-fold) when the doses increased from 3 to 6 mg/kg (1.3-, 1.7-, and 2-fold, respectively). In contrast, the pediatric model displayed linear kinetics with proportional increase in AUCs (1.4-, 1.8-, and 2.3-fold, respectively), (Supplementary Figure 2). Predicted clearance was 3.1 and 6.0 mL/min/kg, which were within 25 and 11 % of the published values of 2.0 and 6.7 mL/min/kg, respectively. Preliminary analysis of oral bioavailability yielded a prediction of 88 % for adults and approximately 100 % for children, showing appropriate prediction for the adults but over-prediction for children. Predictions in the adult and pediatric populations, using Berkeley Madonna, used average physiologic and in vitro parameters, and omitted ranges and standard deviations. Because of this limitation and the unusual preliminary result of predicting nearly double the observed pediatric oral bioavailability, a population-based approach was deemed essential.
3.2 Model Simulations in Healthy Adult Volunteers
Initial population-based model development used standard Simcyp parameters from the healthy adult volunteer simulator. The model was customized for voriconazole (Fig. 2) based on specific hepatic in vitro metabolism and physicochemical properties (Table 1). The pilot simulations tested both intravenous infusions and oral doses as described in Sect. 2. The output demonstrated a biphasic profile indicating a two-compartment model, consistent with published data [4]

Chemical structure of voriconazole as well as the model structure. The model structure defines all organ compartments and blood flow that were used in the simulations. Hashed arrows indicate dosing compartments, including intravenous and oral regimens. Thick black, downwards arrows indicate elimination from kidneys and liver for both the adult and pediatric populations and from the intestines in the pediatric population
Initially, simulations were used to test the model’s capacity to describe voriconazole plasma concentration after multiple intravenous infusion doses in adults. Table 2 lists pharmacokinetic parameters predicted by the model relative to corresponding published values. Model output for intravenous infusions generated a predicted clearance value of 2.4 mL/min/kg (range 1.0–4.8 mL/min/kg), agreeing well with the clinically observed clearance of 2.0 mL/min/kg, a 17 % prediction error. Predicted V d varied from 3.39 to 3.65 L/kg, which differed from the observed values of 4.6 L/kg by 20.7–26.3 %. The prediction error for AUC and C max in adults increased from 2.6 to 82 % and 2.6 to 39.7 %, respectively, as the dose decreased from 5 to 3 mg/kg. This is because of the known inability of the model to simulate non-linearity of metabolism. For the 200-mg oral dose in adults, the predicted AUC and Cmax values corresponded well with observed values (Table 3). The model predicted the mean oral bioavailability of 83 % in adults, which agrees well with the clinically observed mean value of 87 %. The predicted 90 % confidence interval for oral bioavailability was 69–93 %, matching the clinically observed range of 75–96 % [23–25]. Thus, with the exception of AUC prediction at the low dose of 3 mg/kg, the errors for predictions of all pharmacokinetic pararmeters were within the accepted limit of <50 %, indicating the validity of the model.
After validating the predictions of the pharmacokinetic parameters, the plasma concentrations of the intravenous infusion and oral simulations were overlaid on observed data for visual predictive checks. Figure 3a shows plots of plasma concentrations as a function of time for 4 mg/kg dose, yielding good fit to observed plasma concentration. Despite differences in the visual predictive checks for an oral dose of 200 mg, the 90 % confidence interval of predicted plasma concentrations encompassed most of the variability (shown as standard deviation) observed in clinical trials in both dosing regimens (Fig. 3b). The model prediction of the individual plasma concentrations for oral dosing captures the concentrations at the early time points, but not the concentrations at later time points owing to the software’s inability to simulate non-linearity. Despite this, the model accurately predicted the pharmacokinetic parameters, including AUC, C max, and bioavailability, demonstrating that the adult model adequately describes voriconazole in the adult population.

Concentration vs. time profiles for a 4 mg/kg intravenous (infusion) dose and b 200 mg oral dose. The squares represent observed data with the associated standard deviation. The solid line represents the simulated mean concentrations as predicted by the PBPK model for the adult population, and the dashed lines show the simulated 90 % confidence interval. Adult visual predictive checks were based on the dosing regimen reported by Purkins et al. [16, 17]. IV intravenous, PO oral, PBPK physiologically based pharmacokinetic
3.3 Initial Model Simulations in the Pediatric Population
Using a similar approach to the adult model, a pediatric model was created by integrating appropriate in vitro data and physiologic parameters. The pharmacokinetic parameters generated by the pediatric intravenous infusion model are listed in Table 2. The model predicted a clearance of 5.0 mL/min/kg (range of 1.3–12.3 mL/min/kg) compared with the clinically observed clearance of 6.7 mL/min/kg, representing a 25 % error. The value for V d in adults was used, as a pediatric-specific V d value is unavailable. The model predicted the values for AUC and C max that corresponded well to the observed values with an error of 2.5–32.3 % and 1.0–22.4 %, respectively. Simulated AUC of the 8 mg/kg dose was similar to pharmacokinetic parameters of the adult 4 mg/kg dose.
For the oral doses, the predicted AUC and C max were overestimated by more than 100 and 30 %, respectively (Table 3). Furthermore, the model over-predicted the bioavailability by 24–82 %, with the predicted values of 60–94 % within 90 % confidence interval, nearly twofold higher than observed clinical values. The overestimates for AUC and bioavailability suggest that the initial model does not adequately account for the factors that contribute to poor oral bioavailability, e.g., intestinal permeability and/or first-pass metabolism, during the absorption phase.
The visual predictive checks for the pediatric population are shown in Fig. 4a–c and represent the multiple intravenous infusions at doses of 4, 6, and 8 mg/kg, respectively. These profiles show that the simulated mean concentrations for both doses yielded good fit to observed plasma concentrations. Conversely, the calculated profiles for oral 4 and 6 mg/kg doses (Fig. 4d and f, respectively) over-predict plasma concentrations over the entire time period, indicating that the model does not account for all pediatric metabolism and/or absorption barriers, at early or later time points. However, the shapes of the predicted profiles resemble the shape of the profiles for the observed data, suggesting that the absorption parameters are appropriate.

Concentration vs. time profiles for an intravenous dose of a 4 mg/kg, b 6 mg/kg, and c 8 mg/kg, and 4 mg/kg oral dose d without and e with intestinal metabolism (intestinal clearance of 2.7 µL/min/mg), and 6 mg/kg oral dose without f and with g intestinal metabolism (intestinal clearance of 2.7 µL/min/mg). Squares represent the experimental values with the associated standard deviation reported by Walsh et al. [5]. The solid lines represent the simulated mean and the dashed lines show the simulated 90 % confidence interval. IV intravenous, PO oral
3.4 Incorporation of Intestinal Metabolism Improves Predictions of Oral Bioavailability in Children
Because the oral bioavailability of voriconazole in adults is nearly 90 %, it is reasonable to conclude that first-pass metabolism is negligible/absent in this population. Despite accounting for the higher hepatic clearance of voriconazole, the pediatric model was unable to predict lower oral bioavailability of voriconazole compared with adults. Because T max values were within 20 % of the observed data, the absorption rate constant was ruled out as a problem in the model. Therefore, it was hypothesized that the lower oral bioavailability in children is due to intestinal first-pass metabolism. To test the hypothesis, a hypothetical intestinal whole organ clearance was incorporated into the pediatric model. A range of CLint values, based on hepatic V max and K m data from in vitro experiments, were tested. K m values were retained in each simulation because values from adult and pediatric hepatic microsomal in vitro experiments were similar. The initial unchanged V max was subsequently reduced until simulated pharmacokinetic parameters fit the observed ranges (Supplementary Table 1).
Incorporating intestinal clearance into the model reduced prediction errors for AUC by ~70 % and for C max by over 50 %. Most importantly, the predicted values for oral bioavailability decreased from 82 to 51 % for both doses, with a 90 % confidence interval range of 27–76 %. Thus, the predicted bioavailability better corresponded to the clinically observed bioavailability range of 44–66 %. Finally, visual predictive checks of the refined pediatric model, shown in Fig. 4e and g, demonstrate that the simulated concentrations for both oral doses better predict the observed concentrations, with most of the observed data superimposing over the predicted values and the 90 % confidence intervals incorporating observed variability.
4 Discussion
Despite significant progress in understanding the ontogeny of DMEs [9–11, 26], our ability to use this knowledge to predict the pharmacokinetic behavior of drugs is far from adequate. Voriconazole is commonly prescribed in vulnerable pediatric populations despite a lack of information on optimal dosing based on clinical evidence. Limited clinical data available for voriconazole disposition in children demonstrate that the drug is cleared ~3-fold more rapidly and has ~50 % lower oral bioavailability in children compared with adults, with significant inter- and intra-individual variability. This type of information has led to therapeutic dose monitoring in children to achieve a concentration range of 1–5.5 µg/mL, and upward adjustment of doses from 4 mg/kg (adult dose) to 9 mg/kg to reach this range [27, 28]. Notably, these doses are higher than the recommended doses in regulatory documents for voriconazole. This approach must be replaced with a more rigorous approach to achieve safe and effective dosing for pediatric populations.
The allometric scaling performed by Yanni et al. [12] showed that hepatic microsomal data, obtained using tissues from children (aged 2–10 years) and adults, could predict the striking differences in in vivo clearance of voriconazole between these two populations. This work was able to show that differences in contribution and capacity of the enzymes allow children to metabolize voriconazole more efficiently, leading to the increased clearance. However, this work did not provide an explanation for the differences in the oral bioavailability. The PBPK model developed in the present study represents a further refinement by integrating the ontogeny of DMEs with changing physiologic parameters.
We demonstrate the proof of concept for this approach using voriconazole as a model drug because (1) clinical pharmacokinetic data are available for adult and pediatric populations for intravenous and oral doses, (2) the pharmacokinetic behavior in children is different from adults with respect to clearance and oral bioavailability, and (3) the current approaches have not produced a satisfactory dosing strategy for children. In this approach, after confirming the dichotomy of pharmacokinetic behavior between two populations in Berkeley Madonna, Simcyp was used because of the availability of both standardized adult and pediatric populations. Differences in physiology between these two populations were included in Simcyp; therefore, only those parameters reflecting age-related changes in DMEs would be considered.
It has been shown previously that voriconazole is predominantly metabolized by CYP3A4, CYP2C19, and FMO3, all of which are highly expressed in the liver and are localized in the endoplasmic reticulum [29–31]. Therefore, metabolism of voriconazole by hepatic microsomes was an appropriate input parameter for the model. Voriconazole is a lipophilic compound of moderate size with no net charge at physiologic pH values, and thus its translocation across cell membranes should occur via passive diffusion without the assistance of transporters. The simulated output of the PBPK model corresponded with previously published pharmacokinetic data in adult subjects, which determined that voriconazole conforms to a two-compartment model [4]. Furthermore, the calculated AUC, C max, and V d values of intravenous infusions in adult subjects were in agreement with published clinical data demonstrating that the applied physiologic and metabolic characteristics could predict clearances and that the liver is the main organ of elimination. In the multiple oral dosing regimens, pharmacokinetic values were in accordance with those published in the literature. The model was also able to predict bioavailability in adults [7].
However, the bioavailability of voriconazole predicted by the initial pediatric model (60–94 %) was strikingly different from the published values of 45–66 % [5–7], and closer to the high oral bioavailability observed in adults. Multiple clinical trials and population pharmacokinetic analyses have not revealed the underlying mechanism that contributes to the difference in bioavailability between children and adults. Many of these reports implicated an increased ratio of “body mass to liver volume” in children compared with adults as a potential reason for differences in clearance and disposition. In apparent agreement with this hypothesis, the results reported by the Thakker laboratory confirmed that higher hepatic V max conferred an increased clearance [12]. Simple allometric scaling of this work determined that in vitro clearance parameters could accurately determine liver clearance. Consequently, higher hepatic clearance was suspected to be the reason for the decreased bioavailability. The initial pediatric model output included only hepatic metabolism of voriconazole to confirm that an increased V max conferred higher clearance. The model demonstrated that clearance in both adult and pediatric populations agreed with published reports, but it was unable to predict bioavailability in children, even after taking into account the increased hepatic clearance and “liver to body mass” ratio. Because the pharmacokinetic parameters of the intravenous model and the T max of the oral models were accurate, this finding suggested that intestinal first-pass metabolism may contribute to the lower oral bioavailability of voriconazole in the pediatric population.
One of the first reports to demonstrate that intestinal first-pass metabolism by CYP3A4 plays a critical role in overall first-pass metabolism was conducted in a study by Kolars et al. [32]. Prior to this pivotal study, cyclosporine metabolism was widely associated with the liver, but not the intestine [32]. Their evidence, based on studies with anhepatic patients during liver transplant procedure, definitively demonstrated that the intestine can play a major role in first-pass metabolism. Although Walsh et al. speculated that developmental differences in intestinal enzyme activity could be responsible for the decrease in voriconazole bioavailability [5], experimental or theoretical evidence that intestinal first-pass metabolism of voriconazole reduces oral bioavailability in children is lacking. To model the intestinal first-pass metabolism, the apparent V max and K m generated from adult and pediatric human liver microsomes were considered as a starting point to establish a hypothetical intestinal CLint. It is reasonable to assume that voriconazole would have similar affinity for its metabolizing enzymes expressed in different tissues. Therefore, the K m was fixed, but the V max was adjusted to find the appropriate rate of metabolism in the intestines to account for the lower observed bioavailability. After incorporating a hypothetical intestinal metabolism into the pediatric PBPK model, the bioavailability of voriconazole was within the range published in the literature, suggesting that the intestine could play an important role in voriconazole metabolism.
The successful modeling of pediatric oral bioavailability that implicates intestinal first-pass metabolism requires that the source of higher V max of children compared with adults be investigated. According to Fakhoury et al., increased CYP3A and P-gp expression in children allude to a marked difference in the “intestinal pie” compared with adult profiles reported by Paine et al. [33, 34]. To our knowledge, pediatric intestinal enzyme content and activity has not been thoroughly studied. Therefore, abundance data from the adult- and pediatric-simulated populations (n = 400/group) were entered into SigmaPlot (version 10.0; Systat Software) and a histogram with 10 bins was created for hepatic and intestinal CYP3A4 and CYP2C19. A distributional analysis was performed to determine if differences in enzyme content could account for pharmacokinetic differences of voriconazole (Fig. 5). In both populations, CYP3A4 content was higher than CYP2C19 in intestinal and hepatic tissues, and hepatic abundance of both enzymes was higher as compared with the intestine, which is consistent with literature reports. This contrasts with the study revealing that (1) CYP2C19 contributes towards a greater percentage of voriconazole metabolism in children and (2) a trend that pediatric CYP2C19 content was higher compared with adults [12]. Furthermore, despite limited published data on pediatric intestinal CYP2C19 enzyme expression, the distribution plots showed age-dependent differences in intestinal enzyme expression [33, 35]. Clearly, the model and distributional analysis highlight the need to determine both the expression and activity of DMEs in the intestine as a function of age.

Distributional analysis (SigmaPlot) of hepatic and intestinal CYP3A4 and CYP2C19 abundance in adults (dotted line) and children aged 2–12 years (solid line). Panels represent the abundance of a hepatic CYP3A4, b hepatic CYP2C19, c intestinal CYP3A4, and d intestinal CYP2C19. CYP cytochrome P450 enzyme
Although incorporation of the first-pass intestinal metabolism of voriconazole appears to improve the ability of the model to predict the oral bioavailability of the drug in children, it does not rule out a potential role of physiologic factors in bioavailability. As voriconazole is traditionally considered a low extraction compound because of its high bioavailability in adults, blood flow is not expected to cause a large difference in its clearance, which implies that gastric-emptying time and small intestinal transit time may be implicated as important parameters affecting rate of drug absorption and systemic exposure in children [36–39]. A sensitivity analysis was conducted to determine if intestinal physiology could influence voriconazole bioavailability in the models. Figure 6 shows no changes in AUC and C max associated with both a 10-fold increase and a decrease in small intestinal transit time (data not shown for gastric emptying time), suggesting that they do not influence absorption.

Sensitivity analysis for small intestinal transit time in adults and children, showing the effect of alterations in the alpha and beta parameters of the Weibull distribution for adult oral dosing on a AUC and b C max, and for pediatric oral dosing on c AUC, and d C max. AUC area under the plasma concentration-time curve, C max maximum concentration
One limitation was the software’s inability to predict non-linear pharmacokinetic behavior of voriconazole in adults, because V max and K m are not input options. This is because of the requirement of Simcyp to employ whole organ clearance instead of enzyme kinetic parameters. Hence, the model overestimated concentrations at later time points because of the inability to account for changes in metabolic rate as concentrations decrease over time. However, the adult model was accepted because of the model’s ability to accurately predict observed adult pharmacokinetic parameters within 50 % of observed values. For the pediatric model, visual predictive checks showed greater accuracy than the adult model, which is not surprising as voriconazole displays linear pharmacokinetics in children and non-linear pharmacokinetics in adults over the same dose range.
Last, a published population pharmacokinetic model attempted to connect the sparse data from pediatic clinical trials to improve dosing [4]. However in a follow-up clinical trial assessing these dosing recommendations, it was concluded that they were inadequate [3, 40]. The PBPK models presented in this paper are the first to identify a potential underlying cause for the pharmacokinetic differences of voriconazole in children compared with adults.
5 Conclusion
The contribution of this study is to test a novel paradigm in developing a rational strategy for pediatric dosing of drugs extensively cleared by metabolism. By relating in vitro metabolism to clinical data, a “bottom-up” approach to finding doses can be validated and then used to prospectively pick a dose or predict drug-drug interactions [41]. A large number of pediatric clinical trials fail because of inadequate dose selection and lack of response. [42]. Modeling has successfully been used to determine optimal doses and increase the efficacy of pediatric clinical trials [43, 44]. This study’s algorithm provides an appealing approach to modeling pediatric drug disposition that can improve prediction of pharmacokinetic parameters in children, and thus reduce ineffective therapy and the need for costly and uncomfortable therapeutic drug monitoring.
In addition, these models are the first important step in identifying underlying mechanistic reasons for the pharmacokinetic differences seen between adults and children. The true significance of this work lies in the fact that our modeling approach suggests age-dependent differences in intestinal enzymatic composition with a major impact on drug bioavailability. Further, it provides a clear path for future studies to investigate this very interesting possibility that children have a stronger intestinal barrier to oral drug absorption.
References
Steinbach WJ, Benjamin DK. New antifungal agents under development in children and neonates. Curr Opin Infect Dis. 2005;18(6):484–9.
Herbrecht R, Denning DW, Patterson TF, Bennett JE, Greene RE, Oestmann JW, et al. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408–15. doi:10.1056/NEJMoa020191.
Driscoll TA, Yu LC, Frangoul H, Krance RA, Nemecek E, Blumer J, et al. Comparison of pharmacokinetics and safety of voriconazole intravenous-to-oral switch in immunocompromised children and healthy adults. Antimicrob Agents Chemother. 2011;55(12):5770–9. doi:10.1128/AAC.00531-11.
Karlsson MO, Lutsar I, Milligan PA. Population pharmacokinetic analysis of voriconazole plasma concentration data from pediatric studies. Antimicrob Agents Chemother. 2009;53(3):935–44. doi:10.1128/AAC.00751-08.
Walsh TJ, Driscoll T, Milligan PA, Wood ND, Schlamm H, Groll AH, et al. Pharmacokinetics, safety, and tolerability of voriconazole in immunocompromised children. Antimicrob Agents Chemother. 2010;54(10):4116–23. doi:10.1128/AAC.00896-10.
Walsh TJ, Karlsson MO, Driscoll T, Arguedas AG, Adamson P, Saez-Llorens X, et al. Pharmacokinetics and safety of intravenous voriconazole in children after single- or multiple-dose administration. Antimicrob Agents Chemother. 2004;48(6):2166–72. doi:10.1128/AAC.48.6.2166-2172.2004.
Leveque D, Nivoix Y, Jehl F, Herbrecht R. Clinical pharmacokinetics of voriconazole. Int J Antimicrob Agents. 2006;27(4):274–84. doi:10.1016/j.ijantimicag.2006.01.003.
Blake MJ, Castro L, Leeder JS, Kearns GL. Ontogeny of drug metabolizing enzymes in the neonate. Semin Fetal Neonatal Med. 2005;10(2):123–38. doi:10.1016/j.siny.2004.11.001.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology: drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–67. doi:10.1056/NEJMra035092.
Koukouritaki SB, Manro JR, Marsh SA, Stevens JC, Rettie AE, McCarver DG, et al. Developmental expression of human hepatic CYP2C9 and CYP2C19. J Pharmacol Exp Ther. 2004;308(3):965–74. doi:10.1124/jpet.103.060137.
Stevens JC, Hines RN, Gu C, Koukouritaki SB, Manro JR, Tandler PJ, et al. Developmental expression of the major human hepatic CYP3A enzymes. J Pharmacol Exp Ther. 2003;307(2):573–82. doi:10.1124/jpet.103.054841.
Yanni SB, Annaert PP, Augustijns P, Ibrahim JG, Benjamin DK Jr, Thakker DR. In vitro hepatic metabolism explains higher clearance of voriconazole in children versus adults: role of CYP2C19 and flavin-containing monooxygenase 3. Drug Metab Dispos. 2010;38(1):25–31. doi:10.1124/dmd.109.029769.
Weiler S, Fiegl D, MacFarland R, Stienecke E, Bellmann-Weiler R, Dunzendorfer S, et al. Human tissue distribution of voriconazole. Antimicrob Agents Chemother. 2011;55(2):925–8. doi:10.1128/AAC.00949-10.
Basic anatomical and physiological data for use in radiological protection: reference values. A report of age- and gender-related differences in the anatomical and physiological characteristics of reference individuals. ICRP Publication 89. Annal ICRP. 2002;32(3–4):5–265.
Bjorkman S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br J Clin Pharmacol. 2005;59(6):691–704. doi:10.1111/j.1365-2125.2004.02225.x.
Purkins L, Wood N, Ghahramani P, Greenhalgh K, Allen MJ, Kleinermans D. Pharmacokinetics and safety of voriconazole following intravenous- to oral-dose escalation regimens. Antimicrob Agents Chemother. 2002;46(8):2546–53.
Purkins L, Wood N, Kleinermans D, Greenhalgh K, Nichols D. Effect of food on the pharmacokinetics of multiple-dose oral voriconazole. Br J Clin Pharmacol. 2003;56(Suppl 1):17–23.
Jones HM, Gardner IB, Watson KJ. Modelling and PBPK simulation in drug discovery. AAPS J. 2009;11(1):155–66. doi:10.1208/s12248-009-9088-1.
Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies: 1. Mechanism-based prediction of volume of distribution. J Pharm Sci. 2002;91(1):129–56.
Damle B, Varma MV, Wood N. Pharmacokinetics of voriconazole administered concomitantly with fluconazole and population-based simulation for sequential use. Antimicrob Agents Chemother. 2011;55(11):5172–7. doi:10.1128/AAC.00423-11.
Kethireddy S, Andes D. CNS pharmacokinetics of antifungal agents. Expert Opin Drug Metab Toxicol. 2007;3(4):573–81. doi:10.1517/17425225.3.4.573.
Cheeti S, Budha NR, Rajan S, Dresser MJ, Jin JY. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm Drug Dispos. 2012. doi:10.1002/bdd.1830.
Lee S, Kim BH, Nam WS, Yoon SH, Cho JY, Shin SG, et al. Effect of CYP2c19 polymorphism on the pharmacokinetics of voriconazole after single and multiple doses in healthy volunteers. J Clin Pharmacol. 2011. doi:10.1177/0091270010395510.
Scholz I, Oberwittler H, Riedel KD, Burhenne J, Weiss J, Haefeli WE, et al. Pharmacokinetics, metabolism and bioavailability of the triazole antifungal agent voriconazole in relation to CYP2C19 genotype. Br J Clin Pharmacol. 2009;68(6):906–15. doi:10.1111/j.1365-2125.2009.03534.x.
Weiss J, Ten Hoevel MM, Burhenne J, Walter-Sack I, Hoffmann MM, Rengelshausen J, et al. CYP2C19 genotype is a major factor contributing to the highly variable pharmacokinetics of voriconazole. J Clin Pharmacol. 2009;49(2):196–204. doi:10.1177/0091270008327537.
van den Anker JN, Schwab M, Kearns GL. Developmental pharmacokinetics. Handbook Exp Pharmacol. 2011;205:51–75. doi:10.1007/978-3-642-20195-0_2.
Bartelink IH, Wolfs T, Jonker M, de Waal M, Egberts TC, Ververs TT, et al. Highly variable plasma concentrations of voriconazole in pediatric hematopoietic stem cell transplantation patients. Antimicrob Agents Chemother. 2013;57(1):235–40. doi:10.1128/AAC.01540-12.
Doby EH, Benjamin DK Jr, Blaschke AJ, Ward RM, Pavia AT, Martin PL, et al. Therapeutic monitoring of voriconazole in children less than three years of age: a case report and summary of voriconazole concentrations for ten children. Pediatr Infect Dis J. 2012;31(6):632–5. doi:10.1097/INF.0b013e31824acc33.
Hyland R, Jones BC, Smith DA. Identification of the cytochrome P450 enzymes involved in the N-oxidation of voriconazole. Drug Metabol Dispos. 2003;31(5):540–7.
Roffey SJ, Cole S, Comby P, Gibson D, Jezequel SG, Nedderman AN, et al. The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human. Drug Metabol Dispos. 2003;31(6):731–41.
Yanni SB, Annaert PP, Augustijns P, Bridges A, Gao Y, Benjamin DK Jr, et al. Role of flavin-containing monooxygenase in oxidative metabolism of voriconazole by human liver microsomes. Drug Metabol Dispos. 2008;36(6):1119–25. doi:10.1124/dmd.107.019646.
Kolars JC, Awni WM, Merion RM, Watkins PB. First-pass metabolism of cyclosporin by the gut. Lancet. 1991;338(8781):1488–90.
Fakhoury M, Litalien C, Medard Y, Cave H, Ezzahir N, Peuchmaur M, et al. Localization and mRNA expression of CYP3A and P-glycoprotein in human duodenum as a function of age. Drug Metabol Dispos. 2005;33(11):1603–7. doi:10.1124/dmd.105.005611.
Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 “pie”. Drug Metabol Dispos. 2006;34(5):880–6. doi:10.1124/dmd.105.008672.
Johnson TN, Tanner MS, Taylor CJ, Tucker GT. Enterocytic CYP3A4 in a paediatric population: developmental changes and the effect of coeliac disease and cystic fibrosis. Br J Clin Pharmacol. 2001;51(5):451–60.
Berseth CL. Gestational evolution of small intestine motility in preterm and term infants. J Pediatr. 1989;115(4):646–51.
Bisset WM, Watt JB, Rivers RP, Milla PJ. Ontogeny of fasting small intestinal motor activity in the human infant. Gut. 1988;29(4):483–8.
Gupta M, Brans YW. Gastric retention in neonates. Pediatrics. 1978;62(1):26–9.
Heimann G. Enteral absorption and bioavailability in children in relation to age. Eur J Clin Pharmacol. 1980;18(1):43–50.
Friberg LE, Ravva P, Karlsson MO, Liu P. Integrated population pharmacokinetic analysis of voriconazole in children, adolescents, and adults. Antimicrob Agents Chemother. 2012;56(6):3032–42. doi:10.1128/AAC.05761-11.
Rostami-Hodjegan A. Physiologically based pharmacokinetics joined with in vitro-in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther. 2012;92(1):50–61. doi:10.1038/clpt.2012.65.
Benjamin DK Jr, Smith PB, Jadhav P, Gobburu JV, Murphy MD, Hasselblad V, et al. Pediatric antihypertensive trial failures: analysis of end points and dose range. Hypertension. 2008;51(4):834–40. doi:10.1161/HYPERTENSIONAHA.107.108886.
Jadhav PR, Zhang J, Gobburu JV. Leveraging prior quantitative knowledge in guiding pediatric drug development: a case study. Pharm Stat. 2009;8(3):216–24. doi:10.1002/pst.394.
Salazar DE, Song SH, Shi J, Rohatagi S, Heyrman R, Wada DR, et al. The use of modeling and simulation to guide clinical development of olmesartan medoxomil in pediatric subjects. Clin Pharmacol Ther. 2012;91(2):250–6. doi:10.1038/clpt.2011.220.
Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD, et al. A PBPK model to predict disposition of CYP3A-metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. CPT: Pharmacomet Syst Pharmacol. 2012;1:e3–256. doi:10.1038/psp.2012.2.
Acknowledgments
This research was supported by PhRMA and AFPE Pre-Doctoral Fellowships.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
N.R.Z. designed and performed the studies, analyzed the data, and prepared the manuscript. D.R.T. conceived and supervised the study, and prepared the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zane, N.R., Thakker, D.R. A Physiologically Based Pharmacokinetic Model for Voriconazole Disposition Predicts Intestinal First-pass Metabolism in Children. Clin Pharmacokinet 53, 1171–1182 (2014). https://doi.org/10.1007/s40262-014-0181-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-014-0181-y