
Abstract
Background and Objectives
This study implemented pharmacokinetic/pharmacodynamic modelling to support the clinical development of RBP-6000, a new, long-acting, sustained-release formulation of buprenorphine for the treatment of opioid dependence. Such a formulation could offer advantages over existing buprenorphine pharmacotherapy by improving patient compliance and reducing the diversion of the product.
Methods
A population pharmacokinetic model was developed using 36 opioid-dependent subjects who received single subcutaneous doses of RBP-6000. Another pharmacokinetic/pharmacodynamic model was developed using μ-opioid receptor occupancy (µORO) data to predict efficacy of RBP-6000 after repeated doses. It was also assessed how buprenorphine plasma concentrations were correlated with opioid withdrawal symptoms and hydromorphone agonist blockade data from 15 heroin-dependent subjects.
Results
The resulting pharmacokinetic model accurately described buprenorphine and norbuprenorphine plasma concentrations. A saturable maximum effect (E max) model with 0.67 ng/mL effective concentration at 50 % of maximum (EC50) and 91 % E max best described µORO versus buprenorphine plasma concentrations. Linear relationships were found among µORO, withdrawal symptoms and blockade of agonist effects.
Conclusion
Previously published findings have demonstrated µORO ≥70 % is needed to achieve withdrawal suppression and blockade of opioid agonist subjective effects. Model simulations indicated that a 200 mg RBP-6000 dose should achieve 2–3 ng/mL buprenorphine average concentrations and desired efficacy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
This study demonstrated the relationship among buprenorphine plasma concentrations, μ-opioid receptor occupancy (µORO) and blockade of opioid agonist effects. |
A saturable maximum effect (E max) model was established between buprenorphine plasma levels and µORO. The desired buprenorphine activity was achieved at µORO ≥70 %. A buprenorphine plasma concentration of 2 ng/mL is required to achieve a µORO of approximately 70 %. |
This analysis provided new insight into the long-acting pharmacokinetic and pharmacokinetic/µORO profiles of RBP-6000. |
1 Introduction
Opioid addiction is a neurobehavioural syndrome characterized by repeated compulsive seeking and use of an opioid despite adverse social, psychological and/or physical consequences. Opioid addiction is a problem with high costs to individuals, families and society. The use of prescription opioids has tremendously increased in the past decade in the USA (from 174 million in 2000 to 257 million in 2009) because of the widespread availability and variety of prescription opioid products, and changes in treatment paradigms [1]. Opioid abuse, addiction, overdose and other health and social consequences of opioid misuse are taking a rapidly growing toll on individuals and institutions in the USA. It is estimated that 2.2–2.4 million individuals initiate non-medical use of opioids in the USA each year and non-medical opioid use now exceeds use of many conventional street drugs, including cocaine and heroin [2]. Overdose deaths from prescription drugs have exceeded those from street drugs since 2002 and have surpassed traffic accidents as a cause of accidental death [3, 4]. In 2011, over 1,252,500 of 2.5 million emergency department (ED) visits associated with drug abuse or addiction involved illicit drugs, including 258,482 ED visits related to heroin and about 420,040 ED visits related to narcotic pain relievers [5].
Opioid receptors are located in both the central nervous system (CNS) and the periphery. In the CNS, they are found in high concentrations in the limbic system and the spinal cord. The natural ligands for the opioid receptors are a group of neuropeptides known as endorphins. Opioid analgesics mimic the action of these natural ligands, but have a more prolonged action as they are not subject to rapid local metabolism. Three major opioid receptor subclasses have been identified: µ-, κ- and δ-. Buprenorphine is a partial opioid agonist at the µ-opioid receptor, with antagonist properties at the κ-opioid receptor. In contrast to a full agonist, buprenorphine at the µ-opioid receptor has less maximal euphoric effect, and a ceiling on its respiratory depressant effects [6]. By binding to µ-opioid receptors in the brain, buprenorphine reduces craving for opioids and opiate withdrawal symptoms, minimizing the need for opioid-dependent patients to use illicit opiate drugs. For the maintenance treatment of opioid dependence, sublingual buprenorphine or buprenorphine/naloxone is typically given as a single daily dose ranging from 4 to 24 mg per day, with the recommended buprenorphine dosage being 16 mg per day [7].
A major issue in the pharmacological treatment of opioid dependence is the high rate of non-adherence [8, 9]. Currently, there is no approved, parenterally administered, sustained-release buprenorphine product indicated for the treatment of opioid dependence. Such a product could offer advantages over existing buprenorphine pharmacotherapy by improving patient compliance and reducing diversion, abuse and unintended exposure, particularly regarding children. In this respect, a new, sustained-release formulation of buprenorphine, RBP-6000, is being developed for injection by the subcutaneous route. RBP-6000 contains 200 mg/mL of buprenorphine base in the ATRIGEL® Delivery System and provides sustained release of buprenorphine over a minimum of 28 days. Following administration of RBP-6000, day-to-day compliance over the ensuing month would not be a potential issue as it is with existing products that are administered sublingually on a daily basis. Also, since RBP-6000 contains buprenorphine base in a sustained-release delivery system (ATRIGEL®), the safety profile and clinical efficacy of RBP-6000 are expected to be similar to those of sublingually administered buprenorphine and buprenorphine/naloxone treatment.
The primary goal of the present study was to develop a model-based approach to rationally support and justify the dose and dosing regimen of RBP-6000 in phase 2 and 3 trials. For this purpose, a modelling strategy was implemented to characterize the population pharmacokinetics of buprenorphine and norbuprenorphine (a major metabolite), and to assess the relationship between buprenorphine and μ-opioid receptor occupancy (μORO). In addition, the relationship between plasma concentration, μORO, withdrawal symptoms and attenuation (i.e. blockade) of hydromorphone challenge agonist effects was explored. Trial simulations were used for predicting the expected μORO after repeated subcutaneous injections of different doses of RBP-6000 administered once monthly. The model-based approach aimed to determine the RBP-6000 dosage range that is expected to sustain a μORO level of 70 % and to establish the corresponding levels of withdrawal symptom suppression and blockade of the effects of exogenously administered opioids.
2 Methods
2.1 Study Design
The clinical study protocol, informed consent forms and all other appropriate study-related documents were reviewed and approved by an independent and appropriately constituted institutional review board (IRB). The IRB was constituted and operated in accordance with the principles and requirements described in the US Code of Federal Regulations (21 CFR Part 56). The study was conducted in accordance with good clinical practice (GCP) as required by US Food and Drug Administration (FDA) regulations, International Conference on Harmonisation (ICH) guidelines and standard operating procedures for clinical investigation and documentation in force at Reckitt Benckiser Pharmaceuticals Inc. (RBP). Compliance with these requirements also indicated conformity with the ethical principles that have their origins in the Declaration of Helsinki. Informed consent was obtained before a subject was enrolled in the study and prior to the commencement of any protocol-driven activities. The investigator (or designated staff member) met with the subject and explained the study in sufficient detail to permit an informed decision to participate.
The present study was a single-centre, open-label, sequential-cohort, single-ascending-dose study. Thirty-six opioid-dependent (by Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision criteria) subjects were randomized to receive 50, 100 or 200 mg of RBP-6000. Subjects in each cohort received a single subcutaneous dose of RBP-6000 on day 1. On day 1, blood samples for measuring plasma concentrations were drawn at 0.5, 1, 2, 4, 6, 8 and 12 h post-dose, daily on day 2 through day 22, and on days 25, 28, 31, 35, 42, 49, 56, 63, 70, 77, 84, 112, 140 and 150. Human ethylenediaminetetraacetic acid (EDTA)-treated plasma samples were analysed for buprenorphine and norbuprenorphine using a validated liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS) method. Human plasma containing buprenorphine, norbuprenorphine and the internal standards, buprenorphine-D4 and norbuprenorphine-D3, was extracted with an organic solvent mixture after the addition of sodium hydroxide solution (liquid–liquid extraction). After extraction, the extract was evaporated and reconstituted, and an aliquot was injected on a Sciex API 5000 LC–MS/MS equipped with an ultra-performance liquid chromatography column. Quantitation was performed using separate weighted (1/x2 for buprenorphine and 1/x for norbuprenorphine) linear least squares regression analyses generated from fortified plasma calibration standards prepared immediately prior to each run. The method was validated for specificity, linearity, lower limit of quantitation, precision, accuracy, recovery and stability for ranges of 0.0250–5.0 ng/mL for buprenorphine and 0.0200–4.00 ng/mL for norbuprenorphine, based on the analysis of 0.500 mL of plasma. The overall precision for both analytes was better than 6.3 %; the overall accuracy was within ±10.3 %. The recoveries for both analytes and internal standards were above 80 %. The established short-term and long-term stability covered the maximum sample storage time (methods unpublished).
2.2 Population Pharmacokinetic Analysis
All data preparation, summary statistics (mean, median, standard deviation and other measures, as appropriate), logistic regression analysis, reports and graphical display presentation were performed using R version 2.14.1 software [10]. The population pharmacokinetic analysis was conducted using NONMEM version 7.2 software [11]. NONMEM was run in a Windows Vista operating system using the Fortran compiler gfortran version 4.6.0. Diagnostic graphics, exploratory analyses and post-processing of NONMEM outputs were performed using R and Xpose version 4.3 software [12]. The Perl-based software Perl-speaks-NONMEM (PsN) version 3.4.2 was used to perform bootstrapping and visual predictive checks (VPCs) [13].
The first-order conditional estimation (FOCE) with interaction (FOCE-I) method was used for estimating the fixed- and random-effect parameters using a nonlinear mixed-effect approach. Appropriateness of the model was evaluated using various goodness-of-fit criteria, including diagnostic scatter plots, likelihood ratio tests (LRTs) and measures of model stability and adequacy (successful convergence, significant digits, matrix singularity). The results of LRTs were considered statistically significant if decreases in the objective function value (OFV) of nested models were more than 3.84 (P < 0.05, 1 degree of freedom) throughout the model-building process.
The inter-individual variability on all the model parameters was assumed to be log-normally distributed. The residual variability, which comprised, but was not limited to, intra-individual variability, experimental errors, process noise and/or model misspecifications, was modelled using additive, proportional and combined error structures.
2.3 Handling of Outliers
An outlier was defined as an aberrant observation that significantly deviated from the rest of observations in a particular individual and did not refer to a subject as an outlier. The proportion of outliers in a data set should be low and such points may be excluded from the analysis given the potential for these observations to negatively impact the convergence and/or parameter estimates (i.e. which may cause a bias) [14]. Outlier detection was based initially on visual examination of individual and pooled pharmacokinetic profiles. Additionally, data points identified with an absolute conditional weighted residual (|CWRES|) >3 during the initial model-building process were excluded from the analysis. The CWRES are weighted residuals calculated using the FOCE method and have been shown to represent a reliable estimate of the distribution of residuals [15]. Given the theoretical distribution of CWRES, it is expected that 99.73 % of the CWRES should lie within the interval −3 to 3; for this reason, values outside this interval were considered as outliers.
2.4 Base Structural Model Development
Buprenorphine is metabolized primarily by cytochrome P450 3A4 to norbuprenorphine [16]. Buprenorphine undergoes extensive first pass in the liver, thus it is administered sublingually with 50–60 % bioavailability. The population pharmacokinetic model was developed to describe simultaneously the concentrations of buprenorphine and norbuprenorphine.
2.5 Covariate Analysis
Age, sex, race and dose were considered in the covariate analysis. Covariate model-building was a step-wise process consisting of a forward and a backward selection procedure. The LRT was used to evaluate the significance of incorporating or removing fixed effects in the population model based on alpha levels that were set a priori. Initially, each covariate was individually included in the base model. A covariate was retained in the model if a reduction in the OFV was ≥3.84 (χ 2 < 0.05). After defining the full model, the significance of each covariate was tested individually by removing each one from the full model. A covariate was retained in the model if, upon removal, the OFV increased by more than 6.64 points (χ 2 < 0.001).
2.6 Model Evaluation
A non-parametric bootstrap resampling method was used to evaluate the stability and robustness of the final pharmacokinetic model [17]. Resampling with replacement generated 100 bootstrap data sets and the final population pharmacokinetic model was fitted repeatedly to each of the 100 bootstrap data sets. The medians and 95 % confidence intervals of parameters obtained from this step were compared with the final parameter estimates. In addition, a VPC was also performed. Results from the VPC were assessed using graphical comparison of the appropriate 90 % prediction intervals from simulated data with overlaid observed data from the original data set.
2.7 Pharmacodynamics: Pharmacokinetics, μ-Opioid Receptor Occupancy, Opioid Withdrawal Syndrome and Agonist Effects
It is recognized that the medication-assisted treatment of opioid dependence is related to the opioid pharmacotherapy occupying brain µ-opioid receptors. The level of receptor occupancy is expected to mediate the abuse and dependence potential of opioids and to predict clinical efficacy. Specifically, higher medication doses are hypothesized to decrease µ-opioid receptor availability (or ‘binding potential’) and provide agonist replacement that minimizes withdrawal symptoms and prevents the reinforcing, euphoric and other effects of abused opioids resulting in greater clinic attendance [18]. Opioid withdrawal symptoms are the body’s physical response to the absence of the opioid, which include muscle aches, restless anxiety, diarrhoea, abdominal cramping, nausea and vomiting. In clinical trials, subjective opioid withdrawal scales are used to quantify these withdrawal effects. In addition, the blockade of hydromorphone challenge agonist effects is measured by subjective drug-effect assessments, which often employ ratings on visual analogue scales using adjectives that reflect abuse potential such as ‘liking’ or ‘good effect’. These measures are quantitative and exhibit dose–response sensitivity to opioid exposure.
The experimental individual values for buprenorphine plasma concentrations, µORO, opioid withdrawal syndrome and opioid-like agonist effects were provided from two previously published clinical trials by coauthor M.K.G. In trial 1, five heroin-dependent subjects underwent buprenorphine induction from 4 mg/day on day 1 to 16 mg/day by day 7 and were maintained at 32 mg/day for 12 days. On the eighth day of the maintenance period, subjects were challenged with the opioid agonist hydromorphone and subjective drug effects were ascertained, and on day 9, blood samples for the measurement of buprenorphine and norbuprenorphine were collected following buprenorphine administration. On the 10th and 11th days of the maintenance period, opioid withdrawal symptoms were measured prior to buprenorphine administration and 1, 2, 3, 6 and 12 h afterwards. On the 12th and final day of the maintenance period, a positron emission tomography (PET) scan with [11C]-carfentanil was administered 4 h after buprenorphine administration to measure µORO. Subjects were titrated down to the subsequent maintenance periods at buprenorphine doses of 16 mg/day for 12 days, 2 mg/day for 12 days and 0 mg/day for 12 days. During each subsequent maintenance period, subjects underwent the hydromorphone challenge, measurement of opioid withdrawal symptoms and a PET scan [18].
In trial 2, ten heroin-dependent subjects were initially maintained for ≥2 weeks on 16 mg/day buprenorphine given as sublingual tablets. Plasma buprenorphine concentrations, opioid withdrawal symptoms and four hydromorphone challenges (to measure subjective opioid agonist drug effects) or four PET brain scans with [11C]-carfentanil (to measure µORO) were conducted at 4, 28, 52 and 76 h after the last daily buprenorphine dose. In addition to characterizing the relationship between buprenorphine plasma concentration and µORO, the study assessed the relationship between µORO and two key clinical effects—opioid withdrawal syndrome and blockade of hydromorphone agonist subjective drug effects [19].
In both trials, opioid agonist and withdrawal symptoms were assessed by using an Opioid Symptom Questionnaire with 16 agonist and 16 withdrawal scale items. Each item was scored from 0 (‘not at all’) to 4 (‘extremely’), yielding total scores ranging from 0 to 64. Buprenorphine attenuation (blockade) of hydromorphone agonist effects was measured by six visual analogue scale ratings including ‘any drug effect’, ‘high’, ‘good drug effect’, ‘bad drug effect’, ‘stimulated’ and ‘sedated’ [18, 19]. From both trials, whole-brain imaging results were used to calculate µ-opioid receptor availability. The percentage µORO was calculated as (100 minus µ-opioid receptor availability).
3 Results
3.1 Population Pharmacokinetic Modelling
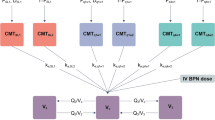
Table 1 presents a demographic summary of the subjects included in the analysis. The analysis data set included 36 subjects for a total of 2,797 observations with 66 observations below the lower limit of quantification. These values were considered as missing in the NONMEM analysis. The buprenorphine and norbuprenorphine measurements were simultaneously fitted using the ADVAN5 TRANS1 routine in NONMEM. The absorption of RBP-6000 from the subcutaneous injection site was described by a dual model that was described by a first-order absorption process associated with the rapid absorption and the first observed peak; and a delayed delivery process that was described by a transit compartment absorption model to mimic the ATRIGEL® Delivery System [20]. The disposition model was a one-compartment model with first-order elimination and first-order conversion to norbuprenorphine. This metabolite was subsequently distributed in a peripheral compartment and eliminated according to a first-order process. Figure 1 presents the final model for buprenorphine and norbuprenorphine.

Final pharmacokinetic model for buprenorphine (BUP) and norbuprenorphine (NOR BUP). In this model, k 12 is the rate of the drug immediately entering into the systemic circulation, k 14 is the rate of the drug entering into the transit compartment system, k k1 is the rate characterizing the delayed process in the transit compartments, k 23 is the rate associated with formation of NOR BUP, k 20 is the elimination rate of BUP, k 30 is the elimination rate of NOR BUP, and k 37 and k 73 are the transfer rate constants between the central and peripheral (Per) NOR BUP compartments, respectively
Initial analysis of the distribution of the CWRES indicated that 28 observations showed an absolute CWRES >3. These values satisfied the definition of outlier measurements. Therefore, a new data set was generated where these measurements were considered as missing observations.
The new analysis data set included 36 subjects for a total of 2,769 observations. The buprenorphine and norbuprenorphine concentrations were again simultaneously fitted using the ADVAN5 TRANS1 routine in NONMEM. The residual error model included a combined additive (Add Err) and proportional components with a different proportional component for buprenorphine (Prop Err BUP) and for norbuprenorphine (Prop Err NorBUP). The results of this analysis were considered as the final model.
Figure 2 presents the goodness-of-fit diagnostic plots and Fig. 3 presents the VPC plots for the final pharmacokinetic model. Overall, there was no apparent bias in the goodness-of-fit diagnostic plots and in the evaluation of the VPCs, suggesting that the final population pharmacokinetic model was adequate in describing the buprenorphine and norbuprenorphine plasma concentration–time courses at RBP-6000 doses of 50 mg, 100 mg and 200 mg.

Goodness-of-fit plots for the final model. a Scatterplot of the individual observed plasma concentrations (ng/mL) versus the individual concentrations (ng/mL). b Scatterplot of the individual observed concentrations (ng/mL) versus the population model-predicted concentrations (ng/mL). c Scatterplot of the absolute individual weighted residuals (|IWRES|) versus the individual concentrations (ng/mL). d Scatterplot of the conditional (Cond.) weighted residuals versus time (hours). The dotted line represents the line of unity

Visual predictive check (VPC) plots for the final pharmacokinetic model enable comparison of statistics derived from the distribution of observations and the distribution of predictions. a VPC plots of buprenorphine (BUP) by dose level. b Magnified VPC plots of BUP by dose level. c VPC plots of norbuprenorphine (Nor-BUP) by dose level. d Magnified VPC plots of Nor-BUP by dose level. The solid line represents the median model prediction, the shaded area represents the 5th and 95th percentiles of the simulated data and the open points are the individual pharmacokinetic measurements
The final population pharmacokinetic parameter estimates for the fixed-effect and random-effect parameters are presented in Table 2 together with the precision of the parameters estimated using the bootstrap procedure. The high level of agreement between the parameter estimated by NONMEM and by the bootstrap procedure, together with the precision of the estimated parameters, supports the adequacy of the model to describe these data.
Empirical Bayesian estimates of individual parameters and random effects were obtained from the base model in the NONMEM analysis. The relationships between individual model parameters and the selected covariates were evaluated graphically. Inspection of the generated plots indicated a potential impact of sex on the volume of distribution for the central norbuprenorphine compartment V3. This hypothesis was formally tested by incorporating sex as a covariate of V3 in the model. However, the resulting objective function did not show a significant change with respect to the base model. Overall, it was not possible to identify any covariate with significant impact on the population pharmacokinetic variability, given the relatively small number of subjects in the study.
3.2 Pharmacodynamic Modelling
A saturable maximum effect (E max) model with an additive error model was used for describing the relationship between buprenorphine plasma concentrations and µORO as presented in equation 1:
where C p is buprenorphine plasma concentration and EC50 is buprenorphine plasma concentration expected to achieve 50 % of the maximal µORO (E max). This model was developed assuming a direct relationship between plasma concentration and µORO without equilibration delay. This model assumes that the metabolite norbuprenorphine has negligible activity with respect to brain µORO. The analysis data set (µORO and buprenorphine pharmacokinetic sampling) included 15 subjects with a total of 59 pharmacokinetic/µORO data. The modelling was performed using the FOCE-I method as implemented in the NONMEM software.
The estimated value for E max (standard error) was 91.4 % (3.94) and the estimated value for EC50 (standard error) was 0.67 (0.19) (ng/mL) (Table 3). The inter-individual variability of E max was not estimated, because of the limited number of measures available in the proximity of the estimated E max value. The adequacy of the final model was evaluated using the VPC method. Four hundred replicates of the original data set were simulated based on the final model, and a 90 % prediction interval was computed based on the simulated data sets. The observed µORO versus the buprenorphine concentration data were plotted on the prediction interval to visually assess the concordance between the simulated and observed data. Statistics of interest including the median were calculated from the simulated and observed data for comparison. The median population prediction and distributions of quantiles (5th, median, 95th) of simulated data were compared graphically with the observed data as shown in Fig. 4a. The graph illustrates the linear relationship between µORO and buprenorphine plasma concentrations up to 2 ng/mL. When buprenorphine levels approached 2–3 ng/mL, the µORO was saturated and reached a plateau with occupancy ranging between 70 and 90 %. A previously published clinical study suggested that the threshold for suppressing withdrawal and blockade of agonist symptom effects is between 50 and 60 % buprenorphine µORO while additional benefit and clinical efficacy was observed at 70 % µORO [19]. As a result of these findings, 70 % µORO was our desired target. The VPCs seems to indicate a larger variability in model predictions compared with observations at the saturation levels (e.g. above 3–4 ng/mL concentrations), and more data would be required to validate the model predictions for that concentration range.

a Visual predictive check plot for the final pharmacokinetic/µ-opioid receptor occupancy (µORO) model for buprenorphine (BUP). The solid line is the median model prediction, the shaded area represents the 5th and 95th percentiles of the simulated data and the open points are the individual µORO measurements. b Observed (diamonds) and model-predicted (solid line) changes in agonist effect following administration of 24 mg hydromorphone (HYD) with respect to µ-opioid receptor (µOR) availability, observed (triangles) and model-predicted (straight dashed line) withdrawal symptoms with respect to µOR availability, and observed (open circles) and model-predicted (curved dash line) buprenorphine plasma concentrations with respect to µOR availability. Changes in agonist effect, withdrawal symptoms and BUP concentrations use the same y axis scale
Regression models were used to describe relationships between mean hydromorphone-induced changes in agonist symptoms, mean withdrawal symptom scores or mean buprenorphine plasma concentrations each with respect to the mean µ-opioid receptor availability (Fig. 4b). These data suggest that a mean buprenorphine plasma concentration of 2 ng/mL is able to provide the desired 70 % μORO. The same conditions are associated with low reported agonist drug effects and withdrawal symptoms (scores ≤2). For the treatment of opioid dependence, the positive clinical outcomes are freedom from withdrawal, cravings and the drug-induced highs and lows of addiction. The individuals who exhibit greater µORO and more suppression of withdrawal symptoms experience better treatment outcomes [21]. As buprenorphine plasma concentrations decline, there is a concomitant increase in subjective hydromorphone agonist drug effects and withdrawal symptoms with a corresponding decrease in µORO.
3.3 Trial Simulation Results: Buprenorphine Exposures after Multiple Subcutaneous Injections of RBP-6000
The simulated drug concentrations of buprenorphine and norbuprenorphine after repeated subcutaneous injections of RBP-6000 were derived from the final model parameter estimates. The 400 hypothetical subjects received four subcutaneous injections of RBP-6000 50, 100, 200 or 300 mg doses separated by 28 days. The objective of this simulation was to predict buprenorphine plasma concentrations after multiple doses of RBP-6000 and to consequently predict the corresponding µORO. The results are presented in Fig. 5. Simulation indicated that the desired ≥70 % receptor occupancy may be achieved after multiple doses of 200 mg RBP-6000.

Simulations of buprenorphine (BUP) plasma concentrations and µ-opioid receptor occupancy (µORO) using the final pharmacokinetic/µORO model. a Model-predicted BUP concentrations following multiple doses once a month at 50, 100, 200 or 300 mg of RBP-6000. The solid line represents the median model prediction and the shaded area represents the 5th and 95th percentiles of the predictions. b Model-predicted median µORO after repeated subcutaneous injections of RBP-6000 administered once a month at doses of 50, 100, 200 or 300 mg
4 Discussion
In this paper we propose a methodological approach that tries to exploit all the information available, using a comprehensive modelling approach to integrate and ‘learn’ from the data generated in different studies of the pharmacokinetic and pharmacokinetic/pharmacodynamic characteristics of RBP-6000. This learning has been subsequently applied to address relevant questions for the clinical development of this compound.
This strategy was implemented by initially defining a population pharmacokinetic model of buprenorphine and norbuprenorphine using data obtained in 36 opioid-dependent subjects who received RBP-6000 with 50, 100 or 200 mg of buprenorphine base. A population pharmacokinetic/µORO model was developed using data (buprenorphine pharmacokinetics and µORO) collected in 15 heroin-dependent subjects (5 receiving buprenorphine daily tablet doses of 32 mg, 16 mg or 2 mg, or placebo, and 10 receiving a buprenorphine daily tablet dose of 16 mg). Finally, the results of the buprenorphine population pharmacokinetic analysis were combined with results of the population pharmacokinetic/µORO analysis to estimate the expected µORO after repeated subcutaneous injections of different doses of RBP-6000 administered once a month. As expected, blockade of hydromorphone agonist effects, withdrawal symptoms and plasma buprenorphine concentrations were correlated with µORO.
Norbuprenorphine is a major metabolite of buprenorphine and a potent agonist of μ-, δ- and κ-opioid receptors. However, while norbuprenorphine is able to bind the µ-opioid receptors, it does not appreciably distribute to the CNS and would not affect our pharmacodynamic endpoints. The reasons why norbuprenorphine was included in the model is that it binds to peripheral µ-opioid receptors, with potential involvement in safety, and is important to our overall clinical development plan. In any case, considering that the norbuprenorphine concentrations were available, it appeared a reasonable strategy to evaluate these data in a comprehensive model for better characterization and understanding of buprenorphine pharmacokinetics.
Analysis of the pharmacokinetic profile of RBP-6000 revealed a complex absorption profile, presenting double peaks and a prolonged plasma terminal half-life. These distinguishing features of the pharmacokinetics of RBP-6000 required the development of a complex pharmacokinetic model accounting for these dual absorption processes: a first absorption process that was associated with an initial rapid delivery from the subcutaneous injection site, and a second absorption process that was associated with a slow release from the ATRIGEL® Delivery System into the systemic circulation. The mean transit time associated with the slow release from the ATRIGEL® Delivery System could be estimated at 10 weeks, which is the likely reason for the curvilinear shape of the plasma concentration–time profile. This model describing the sustained release of RBP-6000 was consistent with the one recently developed to characterize RBP-7000, a long-acting formulation of risperidone that utilizes the same ATRIGEL® Delivery System [22].
The buprenorphine plasma exposure increased proportionally with dose. The established model was stable and described the data well. The covariate analysis was unable to detect any relevant impact of the demographic characteristics of the subjects enrolled in the trial, probably because of the limited sample size.
The clinical efficacy of opioid medication-assisted therapy for the treatment of opioid dependence is believed to result from the medication’s ability to alleviate withdrawal symptoms and bind µ-opioid receptors resulting in blockade of subjective agonist effects. A previously published clinical study suggested that the threshold for suppressing withdrawal and blockade of agonist symptom effects is between 50 and 60 % buprenorphine µORO while additional benefit and clinical efficacy was observed at 70 % µORO [19]. As a result from these previously published findings, the dose selection criterion for the late-phase RBP-6000 drug development was based on the selection of a dose appropriate to reaching and maintaining a µORO greater than 70 % after multiple doses.
The population pharmacokinetic/µORO model fully characterized the relationship between buprenorphine plasma levels and µORO. The relationship between buprenorphine plasma concentration and µORO was best described by an E max model with EC50 of 0.67 ng/mL and E max of 91 %. The E max model showed a linear relationship between µORO up to the desired 70 % receptor occupancy and buprenorphine concentrations up to approximately 2 ng/mL. At buprenorphine concentrations greater than 2 ng/mL, saturation occurred on µORO where a 4.5-fold increase in observed buprenorphine concentrations resulted in observed µORO between 70 % and less than 90 %. Thus, once µORO is saturated, increasing doses are not expected to exert any appreciable effect. A linear correlation was established between buprenorphine clinical efficacy (withdrawal suppression and blockade of hydromorphone agonist subjective effects) and µORO.
Trial simulation indicated that ≥70 % receptor occupancy may be achieved after multiple doses of 200 mg RBP-6000 once every 28 days. Currently, a multiple-ascending-dose study (50–300 mg) of RBP-6000 is ongoing, and the preliminary observed data of buprenorphine plasma concentrations indicated adequate model predictions at each dose level.
5 Conclusion
A comprehensive model-based approach was developed to describe the population pharmacokinetics of RBP-6000 in opioid-dependent subjects and to define the relationships between buprenorphine plasma concentrations, µORO and clinical efficacy. The results of these analyses provided new insight into the long-acting pharmacokinetic and pharmacokinetic/µORO profiles of RBP-6000. These findings indicated that RBP-6000 has the potential to become an effective treatment of opioid dependence by addressing compliance and reducing diversion, abuse and unintended exposure associated with conventional treatments. To our knowledge, this is the first study that empirically combines clinical molecular neuroimaging and plasma concentration and pharmacodynamic data to predict a potentially effective dosing regimen for a novel formulation of an addiction medication, and one of very few studies in the CNS medication development arena to leverage such data. As a general statement of caution, the relatively small sample size of the population enrolled in the pharmacokinetic and µORO studies and the associated variability represent a limitation for generalization of the findings derived from the population pharmacokinetic and population pharmacokinetic/µORO analyses presented here. The modelling and simulation conclusions will be tested in future phase 2 and 3 studies.
References
Katz NP, et al. Prescription opioid abuse: challenges and opportunities for payers. Am J Manag Care. 2013;19:295–302.
Katz NP, et al. Foundations of opioid risk management. Clin J Pain. 2007;23:103–18.
Paulozzi LJ, Ryan GW. Opioid analgesics and rates of fatal drug poisoning in the United States. Am J Prev Med. 2006;31:506–11.
Kochanek KD, Xu J, Murphy SL, Miniño AM, Kung H-C. Deaths: preliminary data for 2009. Natl Vital Stat Rep. 2011;59:1–51.
Substance Abuse and Mental Health Services Administration, Drug Abuse Warning Network (2011). National estimates of drug-related emergency department visits. HHS publication no. (SMA) 13-4760, DAWN series D-39. Rockville: Substance Abuse and Mental Health Services Administration; 2013.
Walsh SL, Preston KL, Stitzer ML, Cone EJ, Bigelow GE. Clinical pharmacology of buprenorphine: ceiling effects at high doses. Clin Pharm Ther. 1994;55:569–80.
SUBOXONE® sublingual film label. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/022410s015lbl.pdf. Accessed 31 Jan 2014.
Weiss RD. Adherence to pharmacotherapy in patients with alcohol and opioid dependence. Addiction. 2004;99:1382–92.
Boothby LA, Doering PL. Buprenorphine for the treatment of opioid dependence. Am J Health-Syst Pharm. 2007;64:266–72.
R Foundation for Statistical Computing (2009). R: a language and environment for statistical computing. http://www.R-project.org. Accessed 14 Dec 2013.
Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user’s guides, 1989–2013. Ellicott City: Icon Development Solutions; 2013.
Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Meth Prog Biomed. 1999;58:51–64.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Meth Prog Biomed. 2005;79:241–57.
Food and Drug Administration (1999). Guidance for industry: population pharmacokinetics. http://www.fda.gov/downloads/Drugs/Guidances/UCM072137.pdf. Accessed 16 Dec 2013.
Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187–97.
Kobayashi K, et al. Human buprenorphine N-dealkylation is catalyzed by cytochrome P450 3A4. Drug Metab Disp. 1998;26:818–21.
Parke J, Holford NH, Charles BG. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput Meth Prog Biomed. 1999;59:19–29.
Greenwald MK, et al. Effects of buprenorphine maintenance dose on mu-opioid receptor availability, plasma concentrations, and antagonist blockade in heroin-dependent volunteers. Neuropsychopharmacology. 2003;28:2000–9.
Greenwald MK, et al. Buprenorphine duration of action: mu-opioid receptor availability and pharmacokinetic and behavioral indices. Biol Psychiatry. 2007;61:101–10.
Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34:711–26.
Greenwald MK, Steinmiller CL. Imaging opioid receptors: applications to substance use disorders. In: Dean R, Negus SS, Bilsky E, editors. Opioid receptors and antagonists: from bench to clinic. New York: Humana Press 2009; p. 45–65.
Gomeni R, Heidbreder C, Fudala PJ, Nasser AF. A model-based approach to characterize the population pharmacokinetics and the relationship between the pharmacokinetic and safety profiles of RBP-7000, a new, long-acting, sustained-release formulation of risperidone. J Clin Pharmacol. 2013;53:1010–9.
Acknowledgments
The authors would like to acknowledge Bradley Vince, DO, President and Medical Director at Vince & Associates (10103 Metcalf Avenue, Overland Park, KS 66212, USA) for patient recruitment in this study.
Conflict of Interest/Disclosure
At the time this manuscript was submitted for publication, A.F. Nasser, C. Heidbreder, P.J. Fudala and B. Zheng were full-time employees of Reckitt Benckiser Pharmaceuticals Inc. M.K. Greenwald was a full-time employee of Wayne State University, and was a paid consultant for Reckitt Benckiser Pharmaceuticals Inc. R. Gomeni was a paid consultant for Reckitt Benckiser Pharmaceuticals Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nasser, A.F., Heidbreder, C., Gomeni, R. et al. A Population Pharmacokinetic and Pharmacodynamic Modelling Approach to Support the Clinical Development of RBP-6000, a New, Subcutaneously Injectable, Long-Acting, Sustained-Release Formulation of Buprenorphine, for the Treatment of Opioid Dependence. Clin Pharmacokinet 53, 813–824 (2014). https://doi.org/10.1007/s40262-014-0155-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-014-0155-0