
Abstract
Background
Erlotinib shows large inter-patient pharmacokinetic variability, but the impact of early drug exposure and genetic variations on the clinical outcomes of erlotinib remains fully investigated. The primary objective of this study was to clarify the population pharmacokinetics/pharmacodynamics of erlotinib in Japanese patients with non-small cell lung cancer (NSCLC). The secondary objective was to identify genetic determinant(s) for the cerebrospinal fluid (CSF) permeability of erlotinib and its active metabolite OSI-420.
Methods
A total of 88 patients treated with erlotinib (150 mg/day) were enrolled, and CSF samples were available from 23 of these patients with leptomeningeal metastases. Plasma and CSF concentrations of erlotinib and OSI-420 were measured by high-performance liquid chromatography with UV detection. Population pharmacokinetic analysis was performed with the nonlinear mixed-effects modelling program NONMEM. Germline mutations including ABCB1 (1236C>T, 2677G>T/A, 3435C>T), ABCG2 (421C>A), and CYP3A5 (6986A>G) polymorphisms, as well as somatic EGFR activating mutations if available, were examined. Early exposure to erlotinib and its safety/efficacy relationship were evaluated.
Results
The apparent clearance of erlotinib and OSI-420 were significantly decreased by 24 and 35 % in patients with the ABCG2 421A allele, respectively (p < 0.001), while ABCB1 and CYP3A5 polymorphisms did not affect their apparent clearance. The ABCG2 421A allele was significantly associated with increased CSF penetration for both erlotinib and OSI-420 (p < 0.05). Furthermore, the incidence of grade ≥2 diarrhea was significantly higher in patients harboring this mutant allele (p = 0.035). A multivariate logistic regression model showed that erlotinib trough (C0) levels on day 8 were an independent risk factor for the development of grade ≥2 diarrhea (p = 0.037) and skin rash (p = 0.031). Interstitial lung disease (ILD)-like events occurred in 3 patients (3.4 %), and the median value of erlotinib C0 levels adjacent to these events was approximately 3 times higher than that in patients who did not develop ILD (3253 versus 1107 ng/mL; p = 0.014). The objective response rate in the EGFR wild-type group was marginally higher in patients achieving higher erlotinib C0 levels (≥1711 ng/mL) than that in patients having lower erlotinib C0 levels (38 versus 5 %; p = 0.058), whereas no greater response was observed in the higher group (67 %) versus the lower group (77 %) within EGFR mutation-positive patients (p = 0.62).
Conclusions
ABCG2 can influence the apparent clearance of erlotinib and OSI-420, and their CSF permeabilities in patients with NSCLC. Our preliminary findings indicate that early exposure to erlotinib may be associated with the development of adverse events and that increased erlotinib exposure may be relevant to the antitumor effects in EGFR wild-type patients while having less of an impact on the tumor response in EGFR mutation-positive patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Background
Erlotinib is a potent epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), and is currently being used to treat patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) [1]. The recommended dose of oral erlotinib (150 mg/day) is the maximum that can be tolerated; thus, an erlotinib dose adjustment is needed to avoid adverse reactions. Since erlotinib shows significant variability in its pharmacokinetics, pharmacokinetic variability can only partly explain differences in outcomes between patients. Previous studies revealed a correlation between erlotinib exposure and the grade of skin toxicity [2–4]. There have been infrequent reports of fatal interstitial lung disease (ILD) in patients receiving erlotinib and gefitinib, another EGFR–TKI [5, 6], and the relationship between high erlotinib concentrations and the occurrence of ILD has been suggested [7–9].
Erlotinib is metabolized in the liver, mainly by cytochrome P450 (CYP) 3A4/3A5 and, to a lesser extent, by CYP1A1/1A2 to produce the active metabolite OSI-420 (desmethyl erlotinib, M14), followed by the formation of many other metabolites including oxidative metabolites (i.e., M2, M12, and M17) and glucuronides (i.e., M10 and M18) [10, 11]. Erlotinib and OSI-420 are considered to be equipotent in inhibiting EGFR tyrosine kinase activity. The CYP3A5*3 allele, resulting in the loss of functional protein, may have the potential to affect the metabolism of erlotinib [12, 13]. It has been demonstrated that erlotinib is transported by the active efflux pumps P-glycoprotein (MDR1/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) [14]. The effect of MDR1 genetic variations on erlotinib pharmacokinetics remains controversial. Thomas et al. [3] reported no influence of MDR1 polymorphisms including 2677G>T/A and 3435C>T on erlotinib disposition, while the ABCB1 1236-2677-3435 TTT haplotype was associated with higher plasma concentrations of erlotinib [15]. On the other hand, there are positive relationships between the ABCG2 421C>A polymorphism, which has been associated with the decreased expression of membrane protein[16], and extensive accumulation of SN-38 [17] and sunitinib [18] in patients who were homozygous for the variant allele, and increased exposure to gefitinib [19] as well as decreased erlotinib disposition [3].
Central nervous system (CNS) metastases remain a frequent complication in NSCLC. Erlotinib can penetrate into the brain and improve CNS metastases [20]. We have previously reported the limited but varying permeability of erlotinib and OSI-420 into the cerebrospinal fluid (CSF) in NSCLC patients who developed leptomeningeal metastases [21]. ABCB1 and ABCG2 are expressed at the blood–brain-barrier (BBB) and prevent the CNS distribution of many drugs including anticancer agents and antiepileptic drugs [22, 23]. However, there is no clinical data on the influence of ABCB1 and ABCG2 polymorphisms on the CSF penetration of erlotinib and OSI-420.
Recent advances in biomarker analysis for the efficacy of gefitinib and erlotinib have identified a number of somatic EGFR mutations, associated with a high sensitivity of tumors to EGFR–TKI [24–26]. The most clinically relevant EGFR mutations are L858R in exon 21 and deletions in exon 19. The objective response rate (ORR) in NSCLC patients with EGFR mutations is reported to be more than 70 %, with median progression-free survival (PFS) ranging from 9 to 13 months [27–32]. However, some patients without EGFR mutations benefit from erlotinib treatment [33, 34]. It has been shown that high plasma drug concentrations were associated with better PFS in NSCLC patients without EGFR mutations treated with gefitinib [35]. The impact of drug exposure on efficacy of erlotinib has not been evaluated considering the EGFR mutation status.
In this study, we aimed to clarify (1) the population pharmacokinetics and pharmacogenomics of erlotinib and its pharmacokinetic and pharmacodynamic relationships including (2) exposure-safety relationship and (3) exposure-efficacy relationship; and to explore (4) genetic determinant(s) for the CNS distribution of erlotinib and OSI-420 in Japanese patients with NSCLC.
2 Patients and Methods
2.1 Patients and Treatment
A total of 88 patients with advanced NSCLC who started erlotinib therapy at Kyoto University Hospital between June 2009 and March 2012 were enrolled (Fig. 1). All patients met the following criteria: histologically confirmed stage IIIb or IV and the presence of clinically and/or radiologically assessable disease. Patients with a performance status ≥2 for whom cytotoxic chemotherapy was considered unacceptable were included. Erlotinib was orally administered at a standard dose of 150 mg/day until progressive disease (PD) or intolerable toxicity. The treatment was continued by dose reductions/interruptions when unacceptable adverse reactions occurred in patients. The demographics and baseline characteristics of patients are summarized in Table 1. This study was performed as part of a treatment and was conducted in accordance with the Declaration of Helsinki and its amendments. The study protocol was approved by the Kyoto University Graduate School and Faculty of Medicine, Ethics Committee. All patients provided written informed consent.

Schematic illustration of the analysis objects in this study. CSF cerebrospinal fluid, NONMEM nonlinear mixed-effects modelling, PD pharmacodynamics, PG pharmacogenomics, PK pharmacokinetics
2.2 Genotyping
Blood DNA was extracted using a Wizard Genomic DNA Purification Kit (Promega KK, Tokyo, Japan). Since the CYP3A4*1B allele is of unknown functional significance, the more clinically relevant CYP3A5*3 (6986A>G) allele was genotyped by polymerase chain reaction (PCR) restriction fragment length polymorphism (RFLT) [36, 37]. Common ABCB1 polymorphisms including 1236C>T, 2677G>T/A, and 3435C>T, and the ABCG2 421C>A polymorphism were examined by direct sequencing or PCR-RFLT [16, 38]. The EGFR mutation status in tumors was obtained from medical records if available. EGFR mutations in exons 18-21 were analyzed by the peptide nucleic acid-locked nucleic acid PCR clamp method [39].
2.3 Pharmacokinetic Study
Serial venous blood samples (2 mL) were collected at 2, 4, 8, and 24 h post-dose on day 1, and at the same time points including before administration on day 8. Trough (C0) blood samplings were also performed whenever ILD was suspected. Plasma was separated by centrifugation (3,000 rpm, 10 min), and stored at −30 °C until the analysis. From 23 patients with CNS metastases (Fig. 1), the remaining part of CSF samples (2 mL), which had been taken before administration for cytologic diagnosis, were collected at a steady state after day 8. Plasma and CSF concentrations of erlotinib and OSI-420 were measured by high-performance liquid chromatography (HPLC) with UV detection [40]. The lower limits of quantification for erlotinib and OSI-420 were both 2 ng/mL. Intra- and inter-assay variabilities were both less than 5 %. Noncompartmental analysis was conducted using WinNonlin version 6.1 software (Pharsight Corporation, Mountain View, CA, USA) to calculate the area under the plasma concentration–time curve over the 24-h dosing interval (AUC24), maximum plasma concentration (Cmax), the time at which Cmax occurred (tmax), minimum plasma concentration (Cmin), and elimination half-life (t½).
2.4 Population Pharmacokinetic Analysis
Population pharmacokinetic analysis was conducted with the nonlinear mixed-effects modelling program (NONMEM) (version 7, double precision, level 1.1; GloboMax, Hanover, MD, USA) using the first-order conditional estimation method with interaction (FOCEI) [41]. A one-compartment model linking a metabolite compartment with first-order absorption and biotransformation to OSI-420 was used to describe the structural pharmacokinetic model for erlotinib (ADVAN5), as shown in Fig. 2. In plasma, OSI-420 was reported to account for only 5 % of the total drug exposure after a single oral dose of erlotinib [10]; thus, the fraction of erlotinib converted to the metabolite OSI-420 (fm) was set at 0.05. For a base model, body weight was used as a covariate to patient body size, along with appropriate exponents, and the relationships of body weight with the apparent clearance and apparent volume of distribution for both erlotinib and OSI-420 were initially tested. These exponents were not significantly different from 1; thus, a linear relationship was chosen for subsequent covariate analysis.

Schematic representation of the structural pharmacokinetic model for erlotinib and its active metabolite OSI-420. f m fraction of erlotinib converted to OSI-420, k a absorption rate constant, k 20 elimination rate constant of erlotinib, k 23 formation rate constant, k 30 elimination rate constant of OSI-420, V 2 apparent volume of distribution of erlotinib, V 3 apparent volume of distribution of OSI-420
The effects of demographic and clinical covariates on pharmacokinetic parameters were examined by forward and backward stepwise regression analysis. For continuous covariates, a linear model and power model centered on a median value were tested for age and clinical laboratory data (albumin, total bilirubin, alanine amino transferase [ALT], aspartate amino transferase [AST], and serum creatinine), respectively. Owing to a lack of an adequate amount of data on plasma drug concentrations during and after the first week of treatment, we could not estimate a linear or nonlinear increase in the apparent oral clearance (CL/F) of erlotinib over time. Instead, the potential influence of a steady state after repetitive dosing on erlotinib CL/F was evaluated by a multiplicative model with an indicator variable (0 or 1). Because the number of patients with a particular genotype of some genes such as ABCG2 and CYP3A5 was small (Table 1), genotype information was included in the population pharmacokinetic model as dichotomous variables, with the typical wild-type genotype as a reference category. The fractional change associated with the categorical covariates including sex and concomitant medication was also examined by a multiplicative model. Because only 3 patients were currently smoking, we did not evaluate smoking history as a covariate. However, given the stimulating effect of smoking on erlotinib clearance [2, 42], we assessed the potential influence of smoking history by comparing the individual Bayesian estimates of erlotinib CL/F between current smokers and non-smokers.
The minimum objective function value (OFV) was used to evaluate the goodness-of-fit of each model with the likelihood ratio test. Upon the inclusion of each covariate, a difference in the OFV (∆OFV) between basic and intermediate models of more than 3.84 was considered significant (p < 0.05, 1 degree of freedom [df]). Upon independent deletion of each covariate from the full regression model, a ∆OFV between full and reduced models of more than 10.83 was required to retain a covariate in the final model (p < 0.001, 1 df). Predictive performance for the final model was assessed by comparing observed and predicted concentrations, as well as by weighted residuals (WRES) versus predicted concentrations and time after dose.
2.5 Evaluation of Safety and Efficacy
All toxicities were graded according to the National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTCAE) version 4.0. The best tumor response was assessed by the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 [43]. Confirmation of complete response (CR) or partial response (PR) was required by means of a second assessment conducted 4 weeks or more after the initial evaluation. Stable disease (SD) was defined as disease control maintained for at least 6 weeks. Long-term efficacy was evaluated by PFS, which was calculated from the date of starting treatment until the date of death or PD, and overall survival (OS). Efficacy analyses were performed on the intent-to-treat (ITT) population. The data cutoff date was 31 August 2012.
2.6 Statistical Analysis
Bivariate correlations were assessed using the Pearson correlation coefficient. A trend for ordered differences of variables across groups was assessed by the Jonckheere–Terpstra test. The cumulative incidence of grade 3/4 adverse events was estimated by adjusting for competing risks (e.g. death or discontinuing treatment due to PD), and differences were evaluated by Gray’s test [44]. A multivariate logistic regression model was applied to determine risk factors for erlotinib-induced adverse events and estimate the adjusted odds ratio (OR) and its 95 % confidence interval (CI). Median PFS and OS were estimated using the Kaplan–Meier method. A multivariate Cox proportional hazards model was used to evaluate the association between PFS or OS and clinical or genomic characteristics and estimate the adjusted hazard ratio (HR) and its 95 % CI. Only those variables with p < 0.20 in the univariate analysis were included in the multivariate analysis. A two-tailed p-value of less than 0.05 was considered significant. All statistical analyses were performed with STATA version 12 (StataCorp LP, College Station, TX, USA).
3 Results
3.1 Exposure to Erlotinib and OSI-420 in Plasma
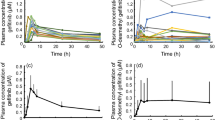
Both erlotinib and OSI-420 exhibited large interindividual variabilities in systemic exposure with a median tmax of 2–4 h (Fig. 3a, b). The median t½ of erlotinib and OSI-420 after the first dose were 32 and 26 h, respectively; thus, a steady state was considered to have been achieved by day 8. The average metabolic ratio of erlotinib to OSI-420 was significantly higher on day 8 than that on day 1 (0.12 versus 0.08; p < 0.0001) (Fig. 3c). Since the sum of the AUC24 of erlotinib and OSI-420 correlated highly with erlotinib C0 levels on day 8 (r 2 = 0.85; p < 0.0001) (Fig. 3d), the C0 level was used as a single predictor for total drug exposure at a steady state. To evaluate drug exposure and the safety/efficacy relationship, patients were classified into three groups based on erlotinib C0 levels: low (quartile 1 [Q1]; n = 21), middle (Q2–Q3; n = 42), and high (Q4; n = 21) (Fig. 3e).

Plasma concentration–time profiles of a erlotinib and b OSI-420 on days 1 and 8. Results are shown as individual data with the mean and standard deviation (SD). c Changes in the metabolic ratio of erlotinib to OSI-420 during the first week. Results are shown as individual data with the mean and SD. The p-value is from a paired t test. d Correlation between erlotinib trough (C0) levels and the sum of AUC24 of erlotinib and OSI-420 on day 8. e Distribution of erlotinib C0 levels on day 8. Vertical lines represent the 25th and 75th percentiles, respectively, dividing the population into 3 groups: low (Q1: 234–843 ng/mL), middle (Q2–Q3: 848–1,684 ng/mL), and high (Q4: 1,711–5,047 ng/mL). AUC 24 area under the concentration–time over 24-h dosing interval
Pharmacogenomic data were available from 86 patients of the population, and the frequencies of germline mutations examined were all in Hardy–Weinberg equilibrium (Table 1). Neither single nucleotide polymorphisms examined in the ABCB1 gene nor the ABCB1 TTT haplotype influenced erlotinib C0 levels on day 8, while the ABCG2 421A allele was significantly associated with increased erlotinib C0 levels (Fig. 4). Although the metabolic ratio of erlotinib to OSI-420 was significantly higher in CYP3A5 expressors versus nonexpressors (p < 0.05), the C0 level was not affected by the CYP3A5 6986A>G polymorphism (Fig. 4f). There were no differences in drug exposure in patients with or without a gastric acid-reducing agent or calcium channel blocker (Table 2). Notably, the Cmin, Cmax, and AUC24 of erlotinib and the Cmin of OSI-420 on day 8 were significantly lower in patients concomitantly treated with antiepileptic drugs (p < 0.05).

Influence of ABCB1 (a, 1236C>T; b, 2677G>T/A; c, 3435C>T; d, 1236-2677-3435 TTT haplotype), e ABCG2 421C>A, and f CYP3A5 6986A>G polymorphisms on steady-state erlotinib trough (C0) levels. Bars indicate the median values. The p-values are from the Kruskal–Wallis test followed by Dunn’s test. CYP3A5 cytochrome P450 3A5, m mutant allele, w wild-type allele
3.2 Population Pharmacokinetic Model
A total of 1284 observations from 86 patients providing pharmacogenomic data were available for population pharmacokinetic analysis (Fig. 1). No significant relationship was observed between clinical laboratory data or concomitant medication and the apparent clearance of erlotinib and OSI-420. The ABCG2 421C>A polymorphism and steady state were identified as significant covariates for the apparent clearance of erlotinib (∆OFV = 42.58 and 128.37, respectively), and the ABCG2 polymorphism was also demonstrated as a significant covariate for that of OSI-420 (∆OFV = 52.56) in univariate analyses. The backward elimination analysis did not exclude any covariates incorporated in the full model (Table 3). The final model and parameter estimates for erlotinib and OSI-420 are summarized in Table 4. The CL/F of erlotinib increased by 34 % after the first week. The ABCG2 421A allele was significantly associated with a 24 % decrease in the CL/F of erlotinib and a 35 % decrease in the apparent clearance of OSI-420. After taking into account all significant covariates in the final population pharmacokinetic model, the interindividual variability in CL/F of erlotinib was reduced from 58.8 % associated with the base model to 44.6 %. In addition, the interindividual variability in CLm/fm of OSI-420 decreased by 10.0 % from 68.3 % associated with the base model. Figure 5 shows the predictive performance for the final model. A small bias was observed in the prediction of OSI-420 concentrations, but the Bayesian method using individual plasma concentration data provided more accurate and less biased predictions for both erlotinib and OSI-420. Furthermore, the WRES for erlotinib as well as OSI-420 were randomly distributed around the null ordinate of perfect agreement throughout predicted concentrations and time after dose. Based on the final model, the average value of the individual Bayesian estimates for erlotinib CL/F in current smokers was approximately 46 % higher than that in non-smoking patients at steady state (6.20 versus 4.24 L/h).

Goodness-of-fit plots for the final model. Observed concentration of erlotinib versus a population predicted concentration and b individual predicted concentration. c Weighted residuals (WRES) for erlotinib versus population predicted concentration and time after dose. Observed concentration of OSI-420 versus d population predicted concentration and e individual predicted concentration. f WRES for OSI-420 versus population predicted concentration and time after dose. The shaded areas show the acceptable range within ±2 units of the null ordinate of perfect agreement
3.3 Cerebrospinal Fluid Penetration of Erlotinib and OSI-420
CSF concentrations of erlotinib were determined in 23 patients (Fig. 1), but those of OSI-420 were available from only 15 patients due to the limit of quantification. Significant correlations were observed between plasma and CSF C0 levels for erlotinib (r 2 = 0.77; p < 0.0001) and OSI-420 (r 2 = 0.28; p = 0.044) (Fig. 6). The average CSF C0 levels of erlotinib and OSI-420 were 36 and 6 ng/mL, respectively, and corresponding plasma concentrations were 1,060 and 138 ng/mL, respectively. The median CSF:plasma concentration ratios of erlotinib and OSI-420 were 3.3 and 3.1 %, respectively.

Correlation between plasma trough (C0) levels of a erlotinib or b OSI-420 and corresponding cerebrospinal fluid (CSF) C0 levels at a steady state after day 8. Influence of ABCB1 and ABCG2 polymorphisms on the CSF to plasma concentration ratio of c erlotinib or d OSI-420. The top, bottom, and line through the middle of a box correspond to the 75th, 25th, and 50th percentiles, respectively. The whiskers on each box extend from the 10th percentile to the 90th percentile. The p-values are from the Mann–Whitney U test. m mutant allele, w wild-type allele
Age, sex, smoking history, and performance status were not associated with the permeability of erlotinib and OSI-420 into the CSF. ABCB1 polymorphisms did not influence the CSF penetration of either erlotinib or OSI-420 (Fig. 6). In contrast, median CSF:plasma concentration ratios were significantly greater in patients with the ABCG2 421A allele than that in wild-type patients (erlotinib, 4.6 versus 3.1 %, p = 0.003; OSI-420, 4.4 versus 2.2 %, p = 0.024).
3.4 Exposure–Safety Relationship
The most common adverse events during erlotinib treatment were skin rash (82 %) and diarrhea (56 %) (Fig. 7a). As for the first required modifications in erlotinib treatment against adverse events, dose reductions and interruptions occurred in 29 (33 %) and 16 (18 %) of the patients, respectively. Treatment discontinuation due to severe adverse reactions occurred in 12 (14 %) of the patients. The most frequent adverse events leading to treatment discontinuation were fatigue and/or anorexia (6.8 %), ILD-like events (3.4 %), liver dysfunction (1 %), and gastrointestinal bleeding (1 %). Severe grade 3/4 toxicities occurred more frequently in patients with increased erlotinib C0 levels on day 8 during the first month (p = 0.007) (Fig. 7b). The development of grade ≥2 skin rash was significantly higher in patients with high erlotinib C0 levels, ABCB1 2677G>T/A, and 3435C>T polymorphisms (Table 5). Multivariate logistic regression analysis showed that only erlotinib C0 levels on day 8 were an independent predictor for developing grade ≥2 skin rash (OR 2.83; 95 % CI 1.10–7.29; p = 0.031). Similarly, the C0 level was independently associated with the development of grade ≥2 diarrhea (OR 3.79; 95 % CI 1.09–13.2; p = 0.037), based on multivariate analysis of sex, body surface area, performance status, erlotinib exposure, and the ABCG2 421C>A polymorphism (Table 5). A positive trend was observed between a higher erlotinib C0 level and higher grade toxicity such as skin, gastrointestinal, and hepatobiliary disorders (Fig. 8). ILD-like events occurred in 3 patients (3.4 %), and the median value of erlotinib C0 levels adjacent to the events was significantly higher than that of patients who did not develop ILD (p = 0.014).

a Drug-related adverse events occurring in 10 % or more of patients by the maximum Common Toxicity Criteria toxicity grade. b Time to first grade 3/4 adverse events during the first month of erlotinib treatment according to the trough (C0) level quartile (Q) on day 8. The p-value is from Gray’s test. ALT alanine aminotransferase, AST aspartate aminotransferase

Associations between erlotinib trough (C0) levels on day 8 and the severity grade of a skin rash, b diarrhea, c oral mucositis, d blood bilirubin increases, e paronychia, and the development of f interstitial lung disease (ILD)-like events. Bars indicate the median values. The p-values are from the Jonckheere–Terpstra test (a–e) or Mann–Whitney U test (f)
3.5 Exposure–Efficacy Relationship
No patient had CR and the disease control rate (CR + PR + SD) of erlotinib for all patients in the ITT population was 66 % (95 % CI 55–76). A significant fraction of patients with EGFR mutations achieved PR (76 %; 95 % CI 59–88), while few wild-type patients had PR (14 %; 95 % CI 4–33) (Fig. 9a). Among EGFR wild-type patients, higher exposure to erlotinib on day 8 (C0 ≥1,711 ng/mL) yielded a higher ORR than that with lower exposure (Q4, 38 % versus Q1–Q3, 5 %; p = 0.058), which was marginally significant, whereas no greater response was achieved in the higher-C0 group (Q4, 67 %) versus the lower-C0 group (Q1–Q3, 77 %) within EGFR mutation-positive patients (p = 0.62) (Fig. 9b). In addition, a more detailed analysis revealed that the middle-C0 group had the highest ORR among the 3 groups (Q1, 67 %; Q2–Q3, 84 %; Q4, 67 %) (Fig. 9c).

a Objective response rate for erlotinib in patients with or without somatic EGFR mutations. b Effect of higher erlotinib exposure on the objective response rate according to the EGFR mutation status. Q1–Q3 and Q4 indicate trough (C0) levels on day 8 of ≤1684 and ≥1711 ng/mL, respectively. The p-values are from Fisher’s exact test. c Objective response rate for erlotinib in patients with somatic EGFR mutations according to erlotinib C0 level quartile (Q) on day 8
The median PFS and OS for all patients in the ITT population were 17.6 and 28.7 weeks, respectively. Multivariate Cox proportional hazards model analysis showed that histology (HR 3.97; 95 % CI 1.15–13.7; p = 0.029), performance status (HR 2.14; 95 % CI 1.12–4.10; p = 0.021), the EGFR mutation (HR 0.48; 95 % CI 0.26–0.91; p = 0.025), and rash grade (HR 0.48; 95 % CI 0.26–0.91; p = 0.025) were independently associated with PFS (Table 6). Furthermore, non-adenocarcinoma (HR 5.07; 95 % CI 1.66–15.5; p = 0.004) and a performance status ≥2 (HR 2.05; 95 % CI 1.07–3.91; p = 0.030) were identified as poor prognostic factors for OS, and the presence of EGFR activating mutations (HR 0.46; 95 % CI 0.23–0.94; p = 0.033) was significantly associated with prolonged OS (Table 6). Neither ABCB1, ABCG2, and CYP3A5 polymorphisms nor early erlotinib exposure were associated with PFS and OS.
4 Discussion
This study demonstrated that the ABCG2 421C>A polymorphism is a significant determinant of interindividual variability in the CNS distribution of erlotinib and OSI-420, as well as in the apparent clearance of both erlotinib and OSI-420 in patients with NSCLC. Our results indicate that higher exposure to erlotinib is associated with higher grade toxicity than lower exposure. Moreover, ILD-like events occurred in patients with extensively elevated C0 levels of erlotinib. These findings suggest that therapeutic drug monitoring (TDM) of plasma erlotinib concentrations may help identify patients at high risk of severe adverse events including ILD.
The mean population pharmacokinetic parameter estimates of erlotinib and OSI-420 were generally consistent with those reported outside Japan [2, 3, 45, 46], which suggests no marked ethnic difference. Kraut et al. [45] reported that there was a significant increase in AUC24, OSI-420/AUC24, erlotinib between day 1 and day 8 in patients with head-neck squamous cell carcinoma. It has been revealed that the metabolism of midazolam in vitro was stimulated after the treatment of liver and intestinal microsomes with erlotinib [11, 47]. Based on these findings, our result showing that the metabolic ratio increased during the first week may be accounted for by the stimulation of metabolism, probably due to the autoinduction and/or activation of CYP3A enzymes by erlotinib. The metabolic ratio was significantly different among CYP3A5*3 genotypes, suggesting that CYP3A5 is involved in the conversion of erlotinib to OSI-420. However, the contribution of this enzyme to the overall CL/F of erlotinib was not significant, which is supported by previous studies [3, 46, 48]. These observations may be explained by the fact that erlotinib is metabolized predominantly by CYP3A4 [11]. Figure 5d shows no structural bias except for a small underestimation of some high concentrations of OSI-420, which were mostly from peak concentrations. One possible explanation for this bias is that the reduced metabolism and/or excretion of OSI-420 may be caused by saturation at high metabolite concentrations in the liver.
Oral absorption of erlotinib is considered to be dependent on gastric pH [49]. Interestingly, no significant pharmacokinetic interaction was observed between erlotinib and gastric acid-reducing agents (Table 2), which is consistent with previous results [48]. It may have been because of their administration in a staggered manner. Moreover, a proton pump inhibitor can inhibit the activity of CYP3A and ABCG2 [50, 51], which may compensate for potential decreases in the oral absorption of erlotinib due to decreased solubility. Based on these results, concomitant medication of gastric acid-reducing agents may not attenuate the effectiveness of erlotinib and should be recommended in patients at high risk of gastrointestinal ulcers [52]. On the other hand, the antiepileptic drugs phenytoin and carbamazepine, which are CYP3A4 inducers, significantly reduced the steady-state Cmax and AUC24 of erlotinib by more than 50 %. Thus, an increased dose of erlotinib should be considered during co-treatment with enzyme-inducing antiepileptic drugs. Both erlotinib and OSI-420 have been identified as substrates for ABCG2 [22]. In addition, ABCB1 may play a role in erlotinib pharmacokinetics and toxicity [15]. We found that the presence of the ABCG2 421A allele resulted in a significant decrease in the apparent clearance of erlotinib (24 %) and OSI-420 (35 %), which is consistent with previous findings [3]. The significant influence of the ABCG2 polymorphism on metabolite clearance indicates that ABCG2 can play a role in the hepatobiliary excretion of OSI-420 in the liver.
In the present study, we could not develop a population pharmacokinetic model describing the interindividual variability for erlotinib and OSI-420 in CSF owing to a limited amount of data on CSF drug concentrations. It has been reported that patient demographics such as age, sex, smoking history, and performance status are not associated with the interindividual variability in the CSF:plasma concentration ratio of gefitinib in patients with NSCLC [53], which is consistent with our results of erlotinib CNS permeability. Although the small sample size of the present study may have limited the detection of an influence of common ABCB1 polymorphisms on the CSF penetration of OSI-420, the ABCG2 421C>A polymorphism was shown to significantly alter the CNS permeability of erlotinib and OSI-420. To our knowledge, this is the first report suggesting that ABCG2 limits the brain distribution of erlotinib and OSI-420 at the BBB in cancer patients. Because of a lower CNS exposure to OSI-420 than erlotinib, it is likely that OSI-420 plays a minor role in the clinical activity of erlotinib for CNS metastases. Based on these findings, higher CSF concentrations may be achieved by high-dose erlotinib administration. Furthermore, despite a similar plasma exposure to erlotinib, its CNS exposure can reach at a higher level in patients with CNS metastases from NSCLC who harbor the ABCG2 421A allele without compromising the safety profile as compared with the wild-type patients. Since ABCG2 expression confers acquired resistance to gefitinib in NSCLC [54], it is of interest to investigate whether the ABCG2 polymorphism can affect its expression at the somatic level in cancer cells and intratumoral concentrations of erlotinib in patients with NSCLC. Future studies should be performed to elucidate the association between CSF concentrations of erlotinib and its antitumor effects towards CNS metastases.
As shown in Fig. 7a, the safety profile of erlotinib in Japanese patients with NSCLC was similar to that observed in previous clinical trials [1, 55]. Recently, increased cumulated drug exposure was shown to be associated with grade 3 or higher toxicity in cancer patients treated with the multikinase inhibitor sorafenib [56]. Similarly, in the present study, increased erlotinib C0 levels on day 8 were significantly associated with a more rapid development of grade 3/4 adverse events and with the occurrence of higher grade toxicity. Multivariate analyses revealed that erlotinib C0 levels on day 8 were a significant independent predictor for grade ≥2 skin rash as well as diarrhea. Our results provide further evidence for the association between the pharmacokinetic variability of erlotinib and skin toxicity [2–4]. In contrast, previous studies showed no significant relationship between erlotinib exposure and diarrhea [2, 3]. This discrepancy may be partly explained by the fact that erlotinib C0 levels on day 8 were highly correlated with total drug exposure (AUC24, erlotinib + OSI-420) in our patient population (Fig. 3d). We also clarified that the ABCG2 421A allele was a potential risk factor for grade ≥2 diarrhea, although this relationship was not significant after adjustments for multiple testing. Our results and similar findings of gefitinib toxicity related to the pharmacogenetics of ABCG2 suggest that diarrhea may evolve from high drug exposure on the gut wall where drug absorption may be enhanced by the ABCG2 421C>A polymorphism [57]. Rudin et al. [46] identified a novel diplotype of two polymorphic loci in the ABCG2 promoter (−15622C>T and 1143C>T) relating to erlotinib-induced diarrhea. These findings indicate that reduced ABCG2 activity due to a genetic variant may predispose patients to gastrointestinal adverse effects from EGFR–TKI.
The pharmacogenetics of sunitinib, a TKI similar to erlotinib, have been reported for toxicity and efficacy [58–60]. The CYP3A5*1 allele was associated with an increased risk of dose reductions [60], as well as with improved PFS in patients with renal cell carcinoma [59]. Furthermore, the ABCB1 and ABCG2 polymorphisms analyzed here showed a tendency towards a worse response to sunitinib [60]. In the present study, however, there was no relationship between these genetic polymorphisms and the efficacy of erlotinib in terms of PFS and OS. These findings suggest that the roles of CYP3A5 and ABCB1/ABCG2 in the treatment outcomes of cancer patients may be different in TKIs and/or among different types of cancer. A higher exposure to erlotinib on day 8 did not contribute to prolonged PFS and OS (Table 6). More patients (n = 8, 19 %) were forced to discontinue treatment due to intolerable toxicities during the first month in the high-C0 group, while only 1 patient (2 %) experienced the early termination of treatment in the low-C0 group. It can be postulated that patients who have been forced to discontinue treatment early due to severe toxicities could not have achieved survival benefits from erlotinib despite high drug exposure.
Previously, the effectiveness and minimal toxicity of erlotinib at a daily dose of 25 mg were shown in NSCLC patients with EGFR mutations [61]. In the present study, lower exposure to erlotinib had a more favorable safety profile than that of higher exposure. Furthermore, the middle range of erlotinib concentrations (Q2–Q3) was associated with a better objective response rate (84 %) than the low (Q1, 67 %) and high (Q3, 67 %) concentrations in patients with EGFR mutations (Fig. 9c). Taking these findings and our previous results [62] into account, patients may benefit from an initial low dose of erlotinib to achieve drug concentrations within the middle range (Q2–Q3, 848–1,684 ng/mL), without any compromise in clinical efficacy, if they have good prognostic factors such as somatic EGFR activating mutations. Additionally, an erlotinib-related skin rash (grade ≥2) was independently associated with improved PFS (Table 6), as reported previously [63]. Dose escalations in erlotinib in relation to the incidence and severity of the rash do not seem to increase its clinical activity [64]. On the other hand, when grade 2 or higher skin rash occurred in patients with elevated erlotinib C0 levels at a steady state, dose reductions to the lowest and most effective level should be considered to avoid unnecessary early treatment discontinuation. Although the number of patients in this study was small, ORR was slightly higher in the higher-C0 group (Q4, 38 %) than in the lower-C0 group (Q1–Q3, 5 %) within a subset of patients with wild-type EGFR (Fig. 9b). Our results imply that higher exposure to erlotinib may be required for more potent inhibition of wild-type EGFR. TDM of imatinib has been suggested to improve clinical outcomes in patients with chronic myeloid leukemia and the target trough level of imatinib has been set above approximately 1,000 ng/mL [65, 66]. However, optimal concentrations of erlotinib have not been identified in the present study. Future studies designed to clarify the therapeutic range of erlotinib in relation to the EGFR mutation status and to prospectively compare the efficacy and safety of erlotinib between pharmacokinetic-guided dosing and fixed dosing should be conducted in a larger number of patients with NSCLC.
5 Conclusions
The ABCG2 421C>A polymorphism can influence the apparent clearance of erlotinib and OSI-420, and their CSF permeabilities in patients with NSCLC. Furthermore, TDM of erlotinib C0 levels at a steady state may be helpful in identifying patients at high risk of severe adverse events including ILD. Higher erlotinib exposure may be relevant to the antitumor effects in EGFR wild-type patients while having less of an impact on the tumor response in patients with EGFR activating mutations.
References
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32.
Lu JF, Eppler SM, Wolf J, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure–safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80:136–45.
Thomas F, Rochaix P, White-Koning M, et al. Population pharmacokinetics of erlotinib and its pharmacokinetic/pharmacodynamic relationships in head and neck squamous cell carcinoma. Eur J Cancer. 2009;45:2316–23.
White-Koning M, Civade E, Geoerger B, et al. Population analysis of erlotinib in adults and children reveals pharmacokinetic characteristics as the main factor explaining tolerance particularities in children. Clin Cancer Res. 2011;17:4862–71.
Hotta K, Kiura K, Takigawa N, et al. Comparison of the incidence and pattern of interstitial lung disease during erlotinib and gefitinib treatment in Japanese patients with non-small cell lung cancer: the Okayama Lung Cancer Study Group experience. J Thorac Oncol. 2010;5:179–84.
Tamura T, Nishiwaki Y, Watanabe K, et al. Evaluation of efficacy and safety of erlotinib as monotherapy for Japanese patients with advanced non-small cell lung cancer (NSCLC); integrated analysis of two Japanese phase II studies. J Thorac Oncol. 2007;2(Suppl 4):S742–3.
Togashi Y, Masago K, Mishima M, et al. A case of radiation recall pneumonitis induced by erlotinib, which can be related to high plasma concentration. J Thorac Oncol. 2010;5:924–5.
Tsubata Y, Hamada A, Sutani A, et al. Erlotinib-induced acute interstitial lung disease associated with extreme elevation of the plasma concentration in an elderly non-small-cell lung cancer patient. J Cancer Res Ther. 2012;8:154–6.
ter Heine R, van den Bosch RT, Schaefer-Prokop CM, et al. Fatal interstitial lung disease associated with high erlotinib and metabolite levels. A case report and a review of the literature. Lung Cancer. 2012;75:391–7.
Ling J, Johnson KA, Miao Z, et al. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab Dispos. 2006;34:420–6.
Li J, Zhao M, He P, et al. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13:3731–7.
Kuehl P, Zhang J, Lin Y, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27:383–91.
Li J, Karlsson MO, Brahmer J, et al. CYP3A phenotyping approach to predict systemic exposure to EGFR tyrosine kinase inhibitors. J Natl Cancer Inst. 2006;98:1714–23.
Marchetti S, de Vries NA, Buckle T, et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1−/−/Mdr1a/1b−/− (triple-knockout) and wild-type mice. Mol Cancer Ther. 2008;7:2280–7.
Hamada A, Sasaki J, Saeki S, et al. Association of ABCB1 polymorphisms with erlotinib pharmacokinetics and toxicity in Japanese patients with non-small-cell lung cancer. Pharmacogenomics. 2012;13:615–24.
Imai Y, Nakane M, Kage K, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Mol Cancer Ther. 2002;1:611–6.
de Jong FA, Marsh S, Mathijssen RH, et al. ABCG2 pharmacogenetics: ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin Cancer Res. 2004;10:5889–94.
Mizuno T, Terada T, Kamba T, et al. ABCG2 421C>A polymorphism and high exposure of sunitinib in a patient with renal cell carcinoma. Ann Oncol. 2010;21:1382–3.
Li J, Cusatis G, Brahmer J, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6:432–8.
Clarke JL, Pao W, Wu N, et al. High dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer. J Neurooncol. 2010;99:283–6.
Togashi Y, Masago K, Masuda S, et al. Cerebrospinal fluid concentration of gefitinib and erlotinib in patients with non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;70:399–405.
Elmeliegy MA, Carcaboso AM, Tagen M, et al. Role of ATP-binding cassette and solute carrier transporters in erlotinib CNS penetration and intracellular accumulation. Clin Cancer Res. 2011;17:89–99.
Lazarowski A, Czornyj L, Lubienieki F, et al. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia. 2007;48(Suppl 5):140–9.
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39.
Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500.
Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–44.
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8.
Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8.
Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57.
Morita S, Okamoto I, Kobayashi K, et al. Combined survival analysis of prospective clinical trials of gefitinib for non-small cell lung cancer with EGFR mutations. Clin Cancer Res. 2009;15:4493–8.
Miller VA, Riely GJ, Zakowski MF, et al. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol. 2008;26:1472–8.
Paz-Ares L, Soulières D, Melezínek I, et al. Clinical outcomes in non-small-cell lung cancer patients with EGFR mutations: pooled analysis. J Cell Mol Med. 2010;14:51–69.
Yoshioka H, Hotta K, Kiura K, et al. A phase II trial of erlotinib monotherapy in pretreated patients with advanced non-small cell lung cancer who do not possess active EGFR mutations: Okayama Lung Cancer Study Group trial 0705. J Thorac Oncol. 2010;5:99–104.
Kobayashi T, Koizumi T, Agatsuma T, et al. A phase II trial of erlotinib in patients with EGFR wild-type advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2012;69:1241–6.
Nakamura Y, Sano K, Soda H, et al. Pharmacokinetics of gefitinib predicts antitumor activity for advanced non-small cell lung cancer. J Thorac Oncol. 2010;5:1404–9.
Phan VH, Tan C, Rittau A, et al. An update on ethnic differences in drug metabolism and toxicity from anti-cancer drugs. Expert Opin Drug Metab Toxicol. 2011;7:1395–410.
Uesugi M, Masuda S, Katsura T, et al. Effect of intestinal CYP3A5 on postoperative tacrolimus trough levels in living-donor liver transplant recipients. Pharmacogenet Genomics. 2006;16:119–27.
Cascorbi I, Gerloff T, Johne A, et al. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001;69:169–74.
Nagai Y, Miyazawa H, Huqun, et al. Genetic heterogeneity of the epidermal growth factor receptor in non-small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid-locked nucleic acid PCR clamp. Cancer Res. 2005;65:7276–82.
Zhang W, Siu LL, Moore MJ, et al. Simultaneous determination of OSI-774 and its major metabolite OSI-420 in human plasma by using HPLC with UV detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;814:143–7.
Beal SL, Sheiner LB. NONMEM User’s Guide. San Francisco: University of California; 1992.
Hamilton M, Wolf JL, Rusk J, et al. Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res. 2006;12:2166–71.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47.
Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509.
Kraut EH, Rhoades C, Zhang Y, et al. Phase I and pharmacokinetic study of erlotinib (OSI-774) in combination with docetaxel in squamous cell carcinoma of the head and neck (SSCHN). Cancer Chemother Pharmacol. 2011;67:579–86.
Rudin CM, Liu W, Desai A, et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol. 2008;26:1119–27.
Dong PP, Fang ZZ, Zhang YY, et al. Substrate-dependent modulation of the catalytic activity of CYP3A by erlotinib. Acta Pharmacol Sin. 2011;32:399–407.
Lind JS, Dingemans AM, Groen HJ, et al. A multicenter phase II study of erlotinib and sorafenib in chemotherapy-naive patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16:3078–87.
Budha NR, Frymoyer A, Smelick GS, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92:203–13.
Moreau C, Debray D, Loriot MA, et al. Interaction between tacrolimus and omeprazole in a pediatric liver transplant recipient. Transplantation. 2006;81:487–8.
Breedveld P, Pluim D, Cipriani G, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65:2577–82.
Kim YH, Masago K, Mishima M. Erlotinib and gastrointestinal ulcer. J Thorac Oncol. 2010;5:1108–9.
Zhao J, Chen M, Zhong W, et al. Cerebrospinal fluid concentrations of gefitinib in patients with lung adenocarcinoma. Clin Lung Cancer. 2013;14(2):188–93.
Chen YJ, Huang WC, Wei YL, et al. Elevated BCRP/ABCG2 expression confers acquired resistance to gefitinib in wild-type EGFR-expressing cells. PLoS One. 2011;6:e21428.
Cohen MH, Johnson JR, Chen YF, et al. FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist. 2005;10:461–6.
Boudou-Rouquette P, Narjoz C, Golmard JL, et al. Early sorafenib-induced toxicity is associated with drug exposure and UGTIA9 genetic polymorphism in patients with solid tumors: a preliminary study. PLoS One. 2012;7:e42875.
Cusatis G, Gregorc V, Li J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–42.
van Erp NP, Eechoute K, van der Veldt AA, et al. Pharmacogenetic pathway analysis for determination of sunitinib-induced toxicity. J Clin Oncol. 2009;27:4406–12.
van der Veldt AA, Eechoute K, Gelderblom H, et al. Genetic polymorphisms associated with a prolonged progression-free survival in patients with metastatic renal cell cancer treated with sunitinib. Clin Cancer Res. 2011;17:620–9.
Garcia-Donas J, Esteban E, Leandro-García LJ, et al. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: a multicentre, observational, prospective study. Lancet Oncol. 2011;12:1143–50.
Yeo WL, Riely GJ, Yeap BY, et al. Erlotinib at a dose of 25 mg daily for non-small cell lung cancers with EGFR mutations. J Thorac Oncol. 2010;5:1048–53.
Togashi Y, Masago K, Fujita S, et al. Differences in adverse events between 250 mg daily gefitinib and 150 mg daily erlotinib in Japanese patients with non-small cell lung cancer. Lung Cancer. 2011;74:98–102.
Pérez-Soler R, Chachoua A, Hammond LA, et al. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. J Clin Oncol. 2004;22:3238–47.
Mita AC, Papadopoulos K, de Jonge MJ, et al. Erlotinib ‘dosing-to-rash’: a phase II intrapatient dose escalation and pharmacologic study of erlotinib in previously treated advanced non-small cell lung cancer. Br J Cancer. 2011;105:938–44.
Picard S, Titier K, Etienne G, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109:3496–9.
Guilhot F, Hughes TP, Cortes J, et al. Plasma exposure of imatinib and its correlation with clinical response in the Tyrosine Kinase Inhibitor Optimization and Selectivity Trial. Haematologica. 2012;97:731–8.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, by a Grant-in-Aid from the Fujiwara Memorial Foundation, and by a Grant-in-Aid from the Nakatomi Foundation. The authors have no conflicts of interest that are directly relevant to the content of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fukudo, M., Ikemi, Y., Togashi, Y. et al. Population Pharmacokinetics/Pharmacodynamics of Erlotinib and Pharmacogenomic Analysis of Plasma and Cerebrospinal Fluid Drug Concentrations in Japanese Patients with Non-Small Cell Lung Cancer. Clin Pharmacokinet 52, 593–609 (2013). https://doi.org/10.1007/s40262-013-0058-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-013-0058-5