
Abstract
Background and Objective
A hydrocodone extended-release (ER) formulation employing the CIMA® Abuse-Deterrence Technology platform was developed to provide resistance against rapid release of hydrocodone when tablets are comminuted or taken with alcohol. This study evaluated the pharmacokinetics of three hydrocodone ER tablet prototypes with varying levels of polymer coating to identify the prototype expected to have the greatest abuse deterrence potential based on pharmacokinetic characteristics that maintain systemic exposure to hydrocodone comparable to that of a commercially available hydrocodone immediate-release (IR) product.
Methods
In this four-period crossover study, healthy subjects aged 18–45 years were randomized to receive a single intact, oral 45-mg tablet of one of three hydrocodone ER prototypes (low-, intermediate-, or high-level coating) or an intact, oral tablet of hydrocodone IR/acetaminophen (APAP) 10/325 mg every 6 h until four tablets were administered, with each of the four treatments administered once over the four study periods. Dosing periods were separated by a minimum 5-day washout. Naltrexone 50 mg was administered to block opioid receptors. Blood samples for pharmacokinetic assessments were collected predose and through 72 h postdose. Parameters assessed included maximum observed plasma hydrocodone concentration (C max), time to C max (t max), and area under the concentration-time curve from time 0 to infinity (AUC0–∞).
Results
Mean C max values were 49.2, 32.6, and 28.4 ng/mL for the low-, intermediate-, and high-level coating hydrocodone ER tablet prototypes, respectively, and 37.3 ng/mL for the hydrocodone IR/APAP tablet; respective median t max values were 5.9, 8.0, 8.0, and 1.0 h. Total systemic exposure to hydrocodone (AUC0–∞) was comparable between hydrocodone ER tablet prototypes (640, 600, and 578 ng·h/mL, respectively) and hydrocodone IR/APAP (581 ng·h/mL). No serious adverse events or deaths were reported. The most common adverse events included headache (26 %) and nausea (18 %).
Conclusion
All three hydrocodone ER tablet prototypes (low-, intermediate-, and high-level polymer coating) demonstrated ER pharmacokinetic characteristics. The hydrocodone ER tablet prototype with the high-level coating was selected for development because of its comparable exposure to the hydrocodone IR/APAP formulation and potentially increased ability to resist rapid drug release upon product tampering because of a higher polymer coating level. All study medications were well tolerated in healthy naltrexone-blocked volunteers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Chronic pain affects approximately 100 million adults in the USA [1]. Opioids have been a mainstay of treatment for patients with moderate to severe pain for years; in 2010, the combination product hydrocodone/acetaminophen (hydrocodone/APAP) was the most commonly dispensed prescription medication in the USA [2]. For a long time, hydrocodone was available in the USA for the treatment of pain only in immediate-release (IR) formulations combined with other medications [3]. US-marketed hydrocodone combination products contain analgesics such as APAP, which can cause liver toxicity when used in high doses over extended periods [4], or aspirin or ibuprofen, which have been associated with complications such as gastrointestinal, renal, cardiovascular, hepatic, and hematologic toxicities [5].
Short-acting opioids (e.g., hydrocodone, oxycodone) typically provide 4–6 h of pain relief and have relatively short plasma half-lives that necessitate frequent dosing. The decreased dosing frequency of extended-release (ER) opioid formulations may offer several advantages, including increased patient compliance and improved pain control, over the more frequent dosing of IR formulations in patients with moderate to severe pain [6, 7]. However, the increased drug load in an ER opioid formulation heightens the need to protect against intentional or accidental dose dumping or the rapid release of active drug. As a result, a number of ER opioids are being developed to deter against the release of large amounts of medication into the body [8, 9]; currently, abuse-deterrent ER opioid formulations for chronic pain are available only for oxycodone and oxymorphone [10].
A new single-agent formulation of hydrocodone ER bitartrate has been developed to provide sustained pain relief with twice-daily dosing. This hydrocodone ER formulation employs the CIMA Abuse-Deterrence Technology (ADT) platform, a novel platform that provides an ER pharmacokinetic profile while protecting against the rapid release of hydrocodone when tablets are comminuted (i.e., broken into small pieces by crushing, milling, grating, or grinding). The ADT platform also provides resistance against intentional and unintentional dose dumping caused by crushing before ingestion, injection, or snorting; chewing; aqueous extraction for inravenous dosing; and alcohol extraction [11]. In the manufacturing process, hydrocodone bitartrate is granulated with a high polymer load and subsequently coated with a polymeric film to ensure controlled release of hydrocodone over an extended period while limiting the release of active medication when tablets are either crushed or exposed to solvents [11]. The polymer-coated granules are compressed into tablets in combination with a gelling matrix that further controls the release of hydrocodone and provides additional resistance against dose dumping when tablets are administered with alcohol (Fig. 1).

The CIMA® Abuse-Deterrence Technology platform manufacturing process
Establishing a balance between the sustained release of hydrocodone and abuse-deterrent characteristics of the hydrocodone ER formulation is an important step in the drug development process. The current exploratory study compared the pharmacokinetics of three hydrocodone ER tablet prototypes with either a relatively low-, intermediate-, or high-level polymer coating and a commercially available formulation of hydrocodone IR/APAP to identify the hydrocodone ER tablet prototype expected to have the greatest abuse deterrence potential based on pharmacokinetic characteristics that maintain systemic exposure to hydrocodone comparable to that of the hydrocodone IR tablet.
2 Subjects and Methods
2.1 Subjects
Eligible subjects were healthy men or women aged 18–45 years with body mass indices of 20–30 kg/m2. Subjects were required to have a negative urine drug screen and alcohol test result. Women had to be either surgically sterile for at least 2 years, 2 years postmenopausal, or, if of child-bearing potential, using a medically acceptable method of contraception during the study and for 30 days after discontinuation of the study drug.
Subjects excluded from this study were those individuals with any clinically significant, uncontrolled medical condition (treated or untreated); a history of alcohol, narcotic, or any other substance abuse (as defined by the Diagnostic and Statistical Manual or Mental Disorders, Fourth Edition, Text Revision) or habitual consumption within the past 2 years of >21 U of alcohol per week (1 U of alcohol equals 1 oz of hard liquor, 5 oz of wine, or 8 oz of beer); clinically significant abnormalities in clinical laboratory values, electrocardiogram, or physical examination findings as determined by the investigator or medical monitor; any disorder that may interfere with medication absorption, distribution, metabolism, or excretion; or use of any systemic or topical prescription or nonprescription medication (except acetaminophen or ibuprofen) within 2 weeks or five half-lives (whichever is longer) before the first dose of study medication. Subjects also were excluded if they had donated >450 mL of blood or had significant blood loss within 56 days before the first dose of study medication; had, after resting 5 min, elevated blood pressure (systolic blood pressure [SBP] >140 mmHg and/or diastolic blood pressure [DBP] >90 mmHg) or low blood pressure (SBP <90 mmHg and/or DBP <45 mmHg); had, after resting 5 min, a pulse <45 or >90 bpm; had, within 2 weeks before the first dose of study drug, a clinically significant consumption of caffeine-containing beverage or food (≥600 mg of caffeine); had used nicotine-containing products within 12 months or had used topical or oral nicotine preparations for smoking cessation within 3 months before the first dose of study drug; or had a known sensitivity or idiosyncratic reaction to hydrocodone, its related compounds, or any metabolites or to naltrexone or acetaminophen. All exclusion criteria were intended to ensure subject safety and prevent the inclusion of subjects with factors that may confound results.
2.2 Study Design
This single-center, randomized, open-label, four-period, crossover study in healthy subjects consisted of a screening visit followed by four open-label treatment periods, a final assessment, and a follow-up visit 48–72 h after final discharge from the study center. Subjects were randomized to receive all of the following four treatments, in random sequence with equal probability, with a minimum 5-day washout between treatment periods: hydrocodone ER 45-mg single intact, oral tablet (Teva Pharmaceuticals, Frazer, PA, USA) with a low-level coating; hydrocodone ER 45-mg single intact, oral tablet with an intermediate-level coating; hydrocodone ER 45-mg single intact, oral tablet with a high-level coating; and a single intact, oral hydrocodone IR/APAP 10/325-mg tablet (Norco®, Watson Pharmaceuticals, Inc., Corona, CA, USA) administered every 6 h until four tablets were administered. A 45-mg dose of hydrocodone ER was chosen because it did not exceed the highest total daily dose of hydrocodone that is currently approved for use in the USA. The hydrocodone IR/APAP 10/325-mg tablet administered every 6 h was chosen as the comparator because it allowed for the assessment of systemic exposure to hydrocodone over a 24-h period after administration of a comparable dose of hydrocodone within the ER and IR formulations.
Study medication was administered at approximately 8 a.m. (±2 h) on the first day of each administration period. For hydrocodone IR/APAP, subsequent doses were administered at approximately 2 p.m. (±2 h) and 8 p.m. (±2 h) on the 1st day and at approximately 2 a.m. (±2 h) on the following day. Subjects were to remain seated during and for 1 h after study medication administration and were not to engage in strenuous exercise during the inpatient periods of the study. Subjects took a single 50-mg tablet of naltrexone with 240 mL of water approximately 15 and 3 h before and 9 and 21 h after study medication administration at 8 a.m. (±2 h) to block opioid receptors and minimize opioid-related adverse events (AEs).
During each hydrocodone ER treatment period, blood samples (3 mL) were collected by venipuncture or indwelling catheter immediately before (approximately 5 min) administration and at 15, 30, and 45 min and 1, 1.25, 1.5, 1.75, 2, 2.25, 2.5, 3, 3.5, 4, 5, 6, 8, 10, 12, 18, 24, 30, 36, 48, 60, and 72 h after each administration of hydrocodone ER. For hydrocodone IR/APAP, samples were collected immediately before (approximately 5 min) administration and at 15, 30, and 45 min and 1, 1.25, 1.5, 1.75, 2, 2.25, 2.5, 3, 3.5, 4, 5, 6, 7, 7.25, 7.5, 12, 13, 13.25, 13.5, 18, 18.25, 18.5, 18.75, 19, 19.25, 19.5, 19.75, 20, 20.25, 20.5, 21, 21.5, 22, 23, 24, 30, 36, 48, 60, and 72 h after study drug administration at 8 a.m. (±2 h).
This study was conducted in full accordance with the Good Clinical Practice: Consolidated Guidance approved by the International Conference on Harmonisation [12] and any applicable national and local laws and regulations. The protocol was approved by the appropriate institutional review board. Written informed consent was obtained from all subjects before study participation.
2.3 Bioanalytical Method
Hydrocodone and hydromorphone (active metabolite) concentrations were measured by PPD (Richmond, VA, USA) using a validated bioanalytical method. The method had intra- and interday precision (coefficients of variation) for pools of quality control samples of ≤15 % other than at the lower limit of quantification, where ≤20 % was acceptable. Both intra- and interday calculated concentrations had to be within 15 % of nominal at all concentrations other than the lower limit of quantification, where up to 20 % deviation from nominal was acceptable. The precision and accuracy of all of the methods used in generating plasma concentration data in these studies exceeded these minimum requirements for assay validation. In addition, stability of the analytes in frozen ethylenediaminetetraacetic acid (EDTA) human plasma was demonstrated for periods exceeding the storage periods of the samples prior to analysis, as well as under all conditions to which study samples or working solutions were subjected.
Levels of hydrocodone and hydromorphone were simultaneously determined using a validated high-performance liquid chromatograpy method with tandem mass spectrometry (HPLC-MS/MS) detection. K2EDTA plasma aliquots (100 µL) were mixed with 25 µL of mixed internal standard working solution (d3-hydrocodone and d3-hydromorphone) and then subjected to supported liquid extraction to isolate the analytes from matrix components. After elution from the solid-phase cartridges, the eluate was evaporated, the samples were reconstituted, and a portion was injected into the HPLC-MS/MS system. Using HPLC with column switching and MS/MS detection using positive ion electrospray, the analytes of interest were separated. The retention times for hydrocodone and hydromorphone were approximately 4.2 and 4.3 min, respectively. The validated quantitation range of the assay was 0.100–100 ng/mL for hydrocodone and 0.0500–50.0 ng/mL for hydromorphone.
2.4 Pharmacokinetic Analyses
The pharmacokinetic parameters assessed for hydrocodone ER and hydromorphone (when feasible), included maximum observed plasma drug concentration (C max); time to C max (t max); area under the plasma drug concentration vs. time curve (AUC) from time 0 to 2 h (AUC0–2); AUC from time 0 to 12 h (AUC0–12); AUC from time 0 to infinity (AUC0–∞); AUC from time 0 to the time of the last measurable plasma drug concentration (AUC0–t ); percentage extrapolation, calculated as 100 × ([AUC0–∞ − AUC0–t ]/AUC0–∞); and terminal elimination half-life (t ½). Based on simulated pharmacokinetic parameters at steady state, fluctuation (%) and swing (%) were also calculated. The same parameters were assessed after administration of the first and last doses of hydrocodone IR/APAP with the exception that t max was calculated relative to the first dose administered. A post hoc analysis also was conducted to evaluate the abuse quotient, defined as C max/t max, for each hydrocodone ER prototype (the larger the ratio, the greater the potential attractiveness for abuse). The abuse quotient for hydrocodone IR/APAP was not calculated given that multiple doses were administered (compared with a single dose for hydrocodone ER).
2.5 Safety and Tolerability
Safety and tolerability were assessed by evaluating AEs, clinical laboratory data, 12-lead electrocardiogram data, physical examination findings, vital signs (pulse, seated blood pressure, body temperature, and respiratory rate), oxyhemoglobin saturation, and use of concomitant medications. AEs were assessed and documented for the study duration (from screening through follow-up 48–72 h after the final discharge from the study center).
2.6 Statistical Analyses
Up to 40 healthy subjects were planned to be enrolled in this study, with the intent that approximately 30 subjects would complete the study. The sample size estimate was not based on statistical considerations. The safety analysis set included all subjects who were randomized to treatment and received at least one dose of study medication. The pharmacokinetic analysis set included all subjects in the safety analysis set who had sufficient data for determining pharmacokinetic parameters relevant to planned comparisons.
Pharmacokinetic parameters and the incidence of AEs were summarized by treatment using descriptive statistics. Between-treatment comparisons were also descriptive, and no formal statistical testing was performed.
Steady-state hydrocodone pharmacokinetic profiles for the three hydrocodone ER tablet prototypes dosed every 12 h and hydrocodone IR/APAP dosed at 4- and 6-h intervals were simulated based on the single-dose data using the nonparametric superposition tool in WinNonlin.
3 Results
3.1 Subjects
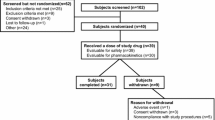
A total of 82 subjects were screened, 40 were enrolled in the study, 39 received at least one dose of study medication, and 37 completed the study (Fig. 2). The 39 subjects who received at least one dose of study medication were included in the safety analysis set, and 36 of the 37 subjects who completed the study had sufficient data to calculate pharmacokinetic parameters for all four study treatments and were included in the pharmacokinetic analysis set. One subject with a previously unreported history of dyspnea discontinued before receiving study medication or naltrexone and was not included in the safety analysis set. Two subjects in the safety analysis set did not complete the study (one had an AE of depressed mood and one withdrew consent). Of note, subjects were permitted to withdraw from a treatment period but continue participation in subsequent periods under certain circumstances. This occurred for one patient who withdrew from the first treatment period because of an AE of vomiting within 2 h after administration of naltrexone; this subject returned for subsequent periods and completed the study but was not included in the pharmacokinetic analysis set as a complete pharmacokinetic database could not be obtained.

Subject disposition
The majority of subjects were male (77 %) and white (64 %). The median age of subjects was 32.0 years (range 23–44 years), and the median body mass index was 25.4 kg/m2 (range 21.4–29.5 kg/m2).
3.2 Pharmacokinetics
The mean plasma hydrocodone concentration vs. time profiles after administration of a single 45-mg dose of the three hydrocodone ER tablet prototypes and four 10-mg doses of hydrocodone IR/APAP through 72 h and through 12 h are shown in Fig. 3. After administration of each of the three hydrocodone ER tablet prototypes, hydrocodone plasma concentrations were maintained throughout the intended 12-h dosing interval. This contrasts with the plasma hydrocodone concentration vs. time profile after administration of hydrocodone IR/APAP, which displayed multiple peaks and troughs over a 24-h period.

Mean (standard deviation) plasma hydrocodone concentration vs. time profiles a through 72 h and b through 12 h in healthy subjects: pharmacokinetic analysis set. APAP acetaminophen, ER extended release, IR immediate release
A summary of pharmacokinetic parameters for the three hydrocodone ER tablet prototypes and hydrocodone IR/APAP are presented in Table 1. C max was inversely correlated with the coating level of the hydrocodone ER tablet prototypes. Compared with hydrocodone IR/APAP, the three hydrocodone ER tablet prototypes had a C max that was approximately 24 % lower to 32 % higher. Although t max occurred much later with the hydrocodone ER tablet prototypes than with hydrocodone IR/APAP (5.9–8.0 vs. 1 h), concentrations near C max for the hydrocodone ER tablet prototypes were achieved early (by ~4 h) and were maintained through approximately 12 h (Fig. 3b). Total systemic exposure to hydrocodone, as assessed by AUC0–∞, was comparable to that of the hydrocodone ER tablet prototypes vs. hydrocodone IR/APAP. As expected, the extent of early release of a drug (as assessed by AUC0–2) with the hydrocodone ER tablet prototypes was inversely correlated with the coating level and was greatest with hydrocodone IR/APAP. Based on a post hoc analysis, the mean (standard deviation) abuse quotient was 8.5 (2.8) for the low-level coating prototype, 4.4 (1.3) for the intermediate-level coating prototype, and 3.6 (1.7) for the high-level coating prototype.
The active metabolite, hydromorphone, was detected after administration of each of the hydrocodone ER tablet prototypes and hydrocodone IR/APAP. Maximum plasma hydromorphone concentration and systemic exposure (AUC0–t ) were approximately 1–2 % of those of hydrocodone after administration of each of the hydrocodone ER tablet prototypes as well as after administration of hydrocodone IR/APAP.
3.2.1 Simulated Steady-State Hydrocodone Pharmacokinetic Profiles
Based on the single-dose data, the anticipated steady-state pharmacokinetic profiles were simulated for the three hydrocodone ER tablet prototypes dosed every 12 h and for hydrocodone IR/APAP dosed at 4- and 6-h intervals in accordance with the recommended therapeutic dosing stated in the package insert [13]. Based on the simulated mean plasma hydrocodone concentrations over a dosing interval at steady state (96 h through 108 h), twice-daily administration of the intermediate- and high-level coating, hydrocodone ER tablet prototypes will produce systemic exposures within the range of exposures anticipated after established therapeutic dose regimens of hydrocodone IR/APAP (Table 2). The intermediate- and high-level coating hydrocodone ER tablet prototypes exhibited a smooth plasma hydrocodone concentration vs. time curve with a single peak during a 12-h dosing period at steady state, in contrast with multiple peaks and troughs for hydrocodone IR/APAP administered every 4 or 6 h (Fig. 4). Fluctuation and swing, respectively, were lower with intermediate-level coating hydrocodone ER (30.3 and 36.2 %) vs. hydrocodone IR/APAP administered every 6 h (53.4 and 72.1 %), comparable to hydrocodone IR/APAP administered every 4 h (29.5 and 34.2 %), and lowest vs. high-level coating hydrocodone ER (24.0 and 27.5 %).

Simulated mean plasma hydrocodone concentration vs. time profiles at steady state for twice-daily administration of the three hydrocodone extended-release (ER) tablet prototypes or administration of the hydrocodone immediate-release (IR)/acetaminophen (APAP) tablet every 4 or 6 h: pharmacokinetic analysis set. The steady-state profiles were simulated using the single-dose data obtained in the current study
3.3 Safety and Tolerability
All 39 subjects in the safety analysis set were administered naltrexone to limit opioid-related AEs. No deaths or serious AEs were reported during the study. Two subjects discontinued from the study because of AEs; one had a previously unreported history of dyspnea and discontinued because of dyspnea before receiving hydrocodone ER or naltrexone, and the other discontinued because of depressed mood after administration of the hydrocodone ER tablet prototype with a high-level coating (the event was mild and considered not related to study medication by the investigator).
All AEs were mild or moderate in intensity. The incidence of common AEs (≥5 %) was generally similar across all four study treatments (Table 3), with a slightly increased frequency of AEs observed with the high-level coating hydrocodone ER tablet prototype. However, the mean plasma hydrocodone concentrations were generally lower with the high-level coating hydrocodone ER tablet prototype compared with the low- and intermediate-coating level prototypes, suggesting the increased frequency is not likely of clinical significance.
Treatment-related AEs (subjects may have reported one or more AE) were observed in three (8 %) subjects after administration of the low-level coating prototype (headache [n = 1]; abdominal pain [n = 2]), one (3 %) subject after administration of the intermediate-level coating prototype (dizziness), five (13 %) subjects after administration of the high-level coating prototype (headache [n = 3]; abdominal pain, nausea, dizziness, diarrhea, and dry mouth [n = 1 each]), and four (11 %) subjects after administration of hydrocodone IR/APAP (nausea, vomiting, and headache [n = 2 each]; constipation, fatigue, and hyperhidrosis [n = 1 each]). Across all study treatments, the most frequently occurring treatment-related AE was headache (n = 6 [15 %]).
Potentially clinically significant reductions in respiratory rate, defined as <10 breaths/min, occurred in a total of 37 subjects during the study. In the majority of the subjects, respiratory rates <10 breaths/min were reported more than once: 16 (42 %) subjects treated with hydrocodone ER with a low-level coating, 20 (53 %) with an intermediate-level coating, 17 (45 %) with a high-level coating, and 17 (46 %) with hydrocodone IR/APAP. Most respiratory rates <10 breaths/min were reported within 4 h of dosing; for 10 subjects, the decreased respiratory rates were reported predose. No AEs potentially associated with decreased respiratory rate were reported. No direct correlation could be made between the measured levels of hydrocodone or hydromorphone and decreased respiratory rate, and no clinically significant decrease in oxygen saturation was reported with a decreased respiratory rate.
No trends or clinically meaningful changes in hematology, chemistry, urinalysis, electrocardiogram, blood pressure, pulse, or oxygen saturation findings were reported.
4 Discussion
Compared with short-acting opioids administered every 4–6 h, extended-release opioids administered once or twice daily are associated with fewer peak-trough fluctuations and thus provide more stable plasma drug concentrations, which may lead to fewer periods of inadequate pain control [7]. In this exploratory, open-label, randomized, crossover study, hydrocodone exposure, based on AUC0–∞, was comparable between all three prototypes of hydrocodone ER tablets (with low-, intermediate-, and high-level polymer coatings) and hydrocodone IR/APAP. Both C max and the extent of the early release of a drug (as assessed by AUC0–2) were inversely correlated with the coating level. t max occurred much later with the hydrocodone ER tablet prototypes (5.9–8.0 h) compared with hydrocodone IR/APAP (1 h); however, concentrations near C max for the hydrocodone ER tablet prototypes were achieved early (by ~4 h) and were maintained through ~12 h. Simulated steady-state profiles indicated that twice-daily administration of the intermediate- and high-level coating hydrocodone ER tablet prototypes would produce sustained plasma hydrocodone concentrations without marked fluctuations over the intended 12-h dosing interval. In contrast, hydrocodone IR/APAP administered every 4 or 6 h produced a concentration vs. time profile with multiple peaks and troughs. Fluctuation (%) and swing (%), respectively, were highest with the low-level coating hydrocodone ER tablet prototype and hydrocodone IR/APAP administered every 6 h, followed by the intermediate-level coating hydrocodone ER tablet prototype and hydrocodone IR/APAP administered every 4 h, and lowest with the high-level coating hydrocodone ER tablet prototype. The prototype with the high-level coating was selected for further development because simulated data suggested that it would maintain systemic exposure to hydrocodone comparable to that of hydrocodone IR/APAP with the least fluctuation, and was the most likely to protect against rapid release of hydrocodone upon product tampering because of its higher level of polymer coating.
Previous studies have shown a correlation between the rate of increase in plasma drug concentrations and the likelihood of drug abuse. In one study, rapid administration of pentobarbital produced significantly greater peak scores for subject-reported feelings of “high”, desire for the drug, and overall drug liking than slow administration [14]. When ER opioid formulations are manipulated, dose dumping or rapid release of a drug is of particular concern as this results in higher maximum drug concentrations and greater euphoria over a shorter period of time [15]. In the current study, the hydrocodone ER tablet prototype with the low-level coating had the highest C max (49.2 ng/mL) but the shortest t max (5.9 h) and the greatest exposure to hydrocodone for the first 2 h after dosing (AUC0–2; 18.6 ng·h/mL), whereas the hydrocodone ER tablet prototype with the high-level coating had the lowest C max (28.4 ng/mL) but a longer t max (8.0 h) and the lowest exposure for the first 2 h (7.4 ng·h/mL). Consistent with these data, post hoc calculations of abuse quotients (C max/t max) demonstrated that the hydrocodone ER tablet prototype with the low-level coating had the highest abuse quotient (8.5), and the prototype with the high-level coating had the lowest abuse quotient (3.6). These findings confirm the appropriateness of selecting the prototype with the highest coating level as the one that would theoretically have the greatest abuse-deterrent properties. However, the actual abuse-deterrent properties of the prototype were not directly assessed in this study. Data regarding the abuse potential of hydrocodone ER assessed in randomized, placebo-controlled, clinical abuse potential studies will be presented separately [16].
All three hydrocodone ER tablet prototypes were generally well tolerated in the current population of healthy volunteers concurrently receiving naltrexone, with no serious AEs or deaths. The safety findings observed were generally consistent with the known safety profiles of hydrocodone [13, 17] and naltrexone [18, 19]. The most common AE was headache (26 %); other common AEs were nausea (18 %), abdominal pain (10 %), and vomiting (10 %). Although most (37/39; 95 %) subjects exhibited decreases in respiratory rate both prior to and after study medication administration that met criteria for potential clinical significance, none of these decreases were associated with AEs. In a separate phase I study of hydrocodone ER 90 mg in naltrexone-blocked subjects, a placebo group was included to allow for comparison of safety parameters in the presence and absence of hydrocodone. Potentially clinically significant, respiratory rate values were reported both before and after administration of ER hydrocodone and placebo with a comparable incidence (data on file, Teva Pharmaceuticals). As a result, the decreased respiratory rate values observed for hydrocodone ER are not considered to be clinically significant. It is possible that procedural variability may have influenced these results.
The current study has several limitations, including use of simulated data for steady-state pharmacokinetic comparisons. Data from multiple-dose studies are needed to appropriately assess the steady-state pharmacokinetic characteristics of hydrocodone ER. Additionally, the safety profile of hydrocodone ER observed in healthy naltrexone-blocked subjects may not be similar to that observed in patients not concurrently receiving naltrexone to block opioid receptors and minimize opioid-related AEs.
5 Conclusion
All three prototypes of hydrocodone ER, with low-, intermediate-, and high-level polymer coatings, demonstrated ER pharmacokinetic characteristics. The hydrocodone ER tablet prototype with a high-level coating was selected for further development because it is expected to produce systemic exposure levels comparable to those seen following administration of the recommended therapeutic dosing regimen for hydrocodone IR/APAP, exhibited the least fluctuation, and has the potential to better protect against the rapid release of a drug upon product tampering.
References
Institute of Medicine Committee on Advancing Pain Research, Care, and Education. Relieving pain in America: a blueprint for transforming prevention, care, education, and research. The National Academies Press. Available at http://books.nap.edu/openbook.php?record_id=13172&page=R1. Accessed July 10 2013.
IMS Institute for Healthcare Informatics. The use of medicines in the United States: review of 2010. IMS Institute for Healthcare Informatics. Available at http://www.imshealth.com/deployedfiles/imshealth/Global/Content/IMS%20Institute/Static%20File/IHII_UseOfMed_report.pdf. Accessed April 2 2013.
The American Society of Health-System Pharmacists. Hydrocodone. Available at http://www.nlm.nih.gov/medlineplus/druginfo/meds/a601006.html. Accessed April 2 2013.
FDA press release. FDA limits acetaminophen in prescription combination products; requires liver toxicity warnings. US Food and Drug Administration. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm239894.htm. Accessed April 2 2013.
Barkin RL. Acetaminophen, aspirin, or ibuprofen in combination analgesic products. Am J Ther. 2001;8(6):433–42.
McCarberg BH, Barkin RL. Long-acting opioids for chronic pain: pharmacotherapeutic opportunities to enhance compliance, quality of life, and analgesia. Am J Ther. 2001;8(3):181–6.
Beaulieu AD, Peloso P, Bensen W, et al. A randomized, double-blind, 8-week crossover study of once-daily controlled-release tramadol versus immediate-release tramadol taken as needed for chronic noncancer pain. Clin Ther. 2007;29(1):49–60.
Khan MF, Gharibo C. Abuse deterrent opioids. Tech Reg Anesth Pain Manag. 2010;14:99–103.
Brennan MJ, Stanos S. Strategies to optimize pain management with opioids while minimizing risk of abuse. PM R. 2010;2(6):544–58.
Pappagallo M, Sokolowska M. The implications of tamper-resistant formulations for opioid rotation. Postgrad Med. 2012;124(5):101–9.
CIMA LABS, Inc. OraGuard™: tamper-deterrent, alcohol-resistant extended release technology. 2012. Available at http://www.cimalabs.com/technology/oraguard. Accessed Mar 31 2014.
International Conference on Harmonisation Working Group. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6 (R1). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; June 10, 1996; Washington, DC. Available at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf. Accessed Mar 1 2011.
Norco (hydrocodone bitartrate and acetaminophen tablets USP) [package insert]. Corona: Watson Pharma Inc.; 2013.
de Wit H, Bodker B, Ambre J. Rate of increase of plasma drug level influences subjective response in humans. Psychopharmacology (Berl). 1992;107(2–3):352–8.
Moorman-Li R, Motycka CA, Inge LD, et al. A review of abuse-deterrent opioids for chronic nonmalignant pain. P T. 2012;37(7):412–8.
Darwish M, Bond M, Tracewell W, et al. Evaluation of the abuse potential of an extended release hydrocodone bitartrate tablet formulated with OraGuard™ technology in non-dependent, recreational opioid users [abstract 395]. J Pain. 2013;14(4 Suppl):S74.
Palangio M, Morris E, Doyle RT Jr, et al. Combination hydrocodone and ibuprofen versus combination oxycodone and acetaminophen in the treatment of moderate or severe acute low back pain. Clin Ther. 2002;24(1):87–99.
Chick J, Anton R, Checinski K, et al. A multicentre, randomized, double-blind, placebo-controlled trial of naltrexone in the treatment of alcohol dependence or abuse. Alcohol Alcohol. 2000;35(6):587–93.
Revia (naltrexone hydrochloride tablet, film coated) [package insert]. North Wales: Teva Women’s Health; 2009.
Acknowledgments
This study was sponsored by Cephalon, Inc., now a wholly owned subsidiary of Teva Pharmaceuticals (Frazer, PA, USA). Writing support was provided by Bina J. Patel, PharmD, of Peloton Advantage, LLC, and funded by Teva Pharmaceuticals. The authors also acknowledge the contributions of Denise D’Andrea and Cathy Shu, MD, PhD, formerly of Teva Pharmaceuticals.
Conflict of interest
MB, WT, PR, and RY are employees of Teva Pharmaceuticals. At the time of this study, MD was an employee of Cephalon, Inc., now a wholly owned subsidiary of Teva Pharmaceuticals (Frazer, PA, USA).
Author information
Authors and Affiliations
Corresponding author
Additional information
At the time of this study, MD was an employee of Cephalon, Inc., now a wholly owned subsidiary of Teva Pharmaceuticals (Frazer, PA, USA).
Rights and permissions
About this article

Cite this article
Darwish, M., Bond, M., Tracewell, W. et al. Pharmacokinetics of Hydrocodone Extended-Release Tablets Formulated with Different Levels of Coating to Achieve Abuse Deterrence Compared with a Hydrocodone Immediate-Release/Acetaminophen Tablet in Healthy Subjects. Clin Drug Investig 35, 13–22 (2015). https://doi.org/10.1007/s40261-014-0244-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-014-0244-8