
Abstract
Background and Objective
Reformulated OxyContin® (oxycodone-HCl controlled release) tablets (ORF) became available in the United States in August 2010. The original formulation of OxyContin® (oxycodone-HCl controlled release) tablets (OC) used a delivery system that did not provide inherent resistance to crushing and dissolving. The objective of this study was to compare the pharmacokinetics, tolerability, and safety of finely crushed ORF tablets, coarsely crushed ORF tablets, and finely crushed OC tablets.
Methods
This randomized, single-blind, single-dose, single-center, six-sequence, triple-treatment, triple-period crossover study enrolled eligible healthy adults (aged 18–55 years inclusive). The study evaluated the pharmacokinetics, tolerability, and safety of intranasally administered ORF, both finely crushed and coarsely crushed, as well as finely crushed OC tablets. Plasma oxycodone concentrations were quantified and analyzed to determine the maximum observed plasma concentration (C max), time to maximum plasma concentration (t max), area under the plasma concentration–time curve from hour 0 to the last measurable plasma concentration (AUClast), and area under the plasma concentration–time curve extrapolated to infinity (AUC∞). The abuse quotient (AQ), calculated as C max/t max, served as an index of the average rate of increase in drug concentration from dosing to t max. Intranasal tolerability rating scales (discomfort, itching, burning, pain, runny nose, and stuffiness) and intranasal endoscopy were conducted. Safety assessments included adverse events, vital signs, pulse oximetry (SpO2), and electrocardiograms.
Results
Of 83 subjects screened and enrolled, 30 were randomized to period 1, with 1 subject subsequently discontinuing due to the subject’s choice. Mean C max values for finely crushed ORF (17.1 ng/mL) and coarsely crushed ORF (15.5 ng/mL) were lower than that for finely crushed OC (22.2 ng/mL). Median t max for finely crushed OC (1.0 h) was shorter than that for either finely crushed ORF (2.0 h) or coarsely crushed ORF (3.0 h). Mean AQ values were approximately 66 and 80 % lower, respectively, for finely crushed ORF and coarsely crushed ORF than that for finely crushed OC. Finely crushed ORF, coarsely crushed ORF, and finely crushed OC demonstrated similar total oxycodone exposures (AUC∞). Insufflation of ORF produced greater nasal discomfort and stuffiness than finely crushed OC, although the latter produced higher runny nose scores. No significant difference was found in other nasal tolerability measures. The overall safety profile was as expected following opioid administration in healthy subjects.
Conclusions
In contrast to OC, both finely and coarsely crushed ORF retained some control of oxycodone release. Reduced C max and increased t max for ORF resulted in lower AQ scores for ORF compared with OC. ORF was associated with greater intranasal irritation than OC. These data suggest that ORF has a lower intranasal abuse potential than OC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Opioid analgesic utilization for pain management has increased significantly since 1998; unfortunately, along with the increase in the legitimate use of opioid medications, there has also been an escalation in the abuse of prescription opioids, particularly abuse of controlled-release formulations that contain higher opioid content [1, 2]. Compared with substances such as cocaine and heroin, prescription opioids are relatively easier to obtain and are perceived as more socially acceptable to abuse. They have also been mentioned as possible “gateway” drugs to other illegal substances [3–5].
Experienced abusers and recreational users commonly employ alternate routes of administration (e.g., injecting and snorting) in abusing oral opioid medications [4, 6, 7]. Data suggest that the incidence of injecting oral oxycodone products among these experienced and recreational abusers ranges from 22 to 59 %, and the incidence of intranasal administration ranges from 45 to 92 % [6, 7]. Abuse via these routes first requires tampering to accelerate the release of the active opioid, which rapidly provides a high blood concentration compared with oral delivery [8]. To reduce abuse and misuse, particularly by injecting and snorting oral opioid analgesics, pharmaceutical manufacturers have begun to develop tamper-deterrent products [2].
Reformulated OxyContin® (oxycodone-HCl controlled release; ORF) was developed with the specific intent of reducing abuse and misuse of the original formulation of OxyContin® (OC) without affecting legitimate use for the management of pain. ORF tablets became available in the United States in August 2010 at the same time that shipments of OC were discontinued. OC used a delivery system that did not provide inherent resistance to breaking, crushing, and dissolving. ORF was designed to be orally bioequivalent to OC, but with abuse-deterrent properties that make abuse via nonoral routes of delivery more difficult. During formulation development, laboratory-based tamper testing indicated that, when crushed, ORF tablets fractionate into large pieces that discourage intranasal abuse, and when dissolved in small amounts of aqueous solution, the particles form a viscous solution that discourages intravenous abuse [9].
This study focuses on intranasal administration of oxycodone, because it is associated with faster absorption than oral administration and may therefore pose a greater risk to the abuser [10]. Additionally, intranasal administration involves crushing or pulverizing the tablets, which may compromise their controlled-release properties. It was hypothesized that upon intranasal administration, the tamper-deterrent properties of ORF relative to OC would translate into a reduction in maximum observed plasma concentration (C max), an increase in time to maximum plasma concentration (t max), and a corresponding decrease in the abuse quotient (AQ) [6, 11, 12], suggestive of a decrease in abuse potential.
The objective of this study was to compare the pharmacokinetics, tolerability, and safety of finely crushed ORF tablets, coarsely crushed ORF tablets, and finely crushed OC tablets, each intranasally administered as single 10-mg doses.
2 Methods
2.1 Study Design
This was a randomized, single-blind, single-dose, single-center, six-sequence, triple-treatment, triple-period crossover study designed to evaluate the pharmacokinetics, tolerability, and safety of intranasally administered ORF, both finely crushed (10 mg) and coarsely crushed (10 mg), as well as finely crushed OC (10 mg). Coarsely crushed OC was not tested because simple crushing of OC readily produces a fine powder. To minimize variability, tampered tablets were prepared using standardized equipment and techniques.
Study duration was up to 42 days; a 28-day screening phase was followed by a 7-day treatment phase, with follow-up 3–7 days after the last dose of study drug. The treatment phase was divided into three periods (days 1–3, 3–5, and 5–7). The assignment of subjects to treatment sequence was single-blinded. Efforts were made to prevent subjects from making side-by-side comparisons of study drugs, which were similar, but not identical, in appearance.
This study protocol and its informed consent form was submitted to the Western Institutional Review Board (IRB) for review and approval. It was conducted in accordance with regulatory guidelines and the applicable International Conference on Harmonization guidelines of Good Clinical Practice, as consistent with the Declaration of Helsinki. All subjects provided oral and written consent before conduct of any protocol-related procedures. Subjects were informed that they could discontinue the study at any time.
2.2 Subjects
Subjects were healthy men and women (aged 18–55 years inclusive) with no clinically significant medical history or any other concerns that would have jeopardized the study’s safety or validity, as determined by the principal investigator. Special preference for study enrollment was given to recreational drug users who had experience with opioid use on at least five occasions, and to subjects who reported at least three occasions of intranasal opioid use for the purpose of abuse/misuse within the past year. Subjects were excluded if they had significant obstruction of either naris, or clinically important changes in the intranasal cavity that would interfere with the study procedures or data integrity or compromised the safety of the subject. Subjects with piercings through the nose or a perforated nasal septum were given a lower priority than those without these findings.
Other conditions warranting exclusion from participation were use of any medication/herbal product [except paracetamol (acetaminophen) (2 g/day), vitamin or mineral supplements, birth control, and hormone replacement] within 7 days of study drug administration or for the study duration.
2.3 Treatment
A physical examination, biochemistry tests, and urinalysis were conducted for each subject at screening. Subjects abstained from consuming alcoholic beverages for 48 h prior to initial study drug administration (day 1) and for the duration of the study (i.e., through to the end of study procedures). They were confined to a nonsmoking study facility the day prior to administration of the study drug and throughout all three periods of the study. Subjects abstained from caffeine or xanthine entirely during confinement. While confined at the study site, they received meals at scheduled times that did not conflict with other study-related activities. The same standard meal timing was employed over corresponding study days in each period, but the content of meals varied.
Subjects were randomized to a treatment sequence on the morning of day 1 and received treatments on days 1, 3, and 5 of the treatment phase, with a minimum washout period of 48 h between dose administrations. For each treatment period, subjects completed a 10-h overnight fast before sitting in an upright position and intranasally insufflating a dose of finely crushed ORF (10 mg), coarsely crushed ORF (10 mg), or finely crushed OC (10 mg) through a short thin straw. Dosing was carefully observed by the site staff, and the weight of any drug not successfully insufflated during the 5-min administration period was recorded, with significant amounts of nonadministered drug potentially resulting in subject discontinuation from the study. Subjects continued fasting for 4 h subsequent to dosing and were to remain upright, unless a procedure required that they be in the supine position. They remained confined to the study site throughout all three treatment periods and abstained from strenuous exercise or physical exertion. Treatment procedures were identical for all three treatment periods, with subjects receiving single intranasal doses of study drug in alternating nares (e.g., left-right-left). Telephone follow-ups were conducted 3–7 days after the last dose or following early discontinuation.
2.4 Pharmacokinetic Measures
During each treatment period, blood samples for determining oxycodone plasma concentrations were obtained for each subject just prior to dosing and at 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0, 6.0, 8.0, 10, 12, 16, 24, 28, 32, 36, and 48 h postdose. For each sample, venous blood (4 mL) was drawn via an indwelling catheter or direct venipuncture into tubes containing potassium ethylenediaminetetraacetic acid anticoagulant. A validated liquid chromatography tandem mass spectrometric method quantified plasma concentrations of oxycodone (lower limit of quantitation was 0.1 ng/mL).
Relative bioavailability was determined by comparison to the reference treatment (finely crushed OC). Pharmacokinetic metrics were calculated, whenever possible, based on the plasma concentrations of oxycodone according to the model independent approach. Pharmacokinetic calculations were performed using WinNonLin (Pharsight Corporation, St. Louis, MO, USA; Version 5.2). Noncompartmental analysis of plasma oxycodone concentrations generated values for: maximum observed plasma concentration (C max), time to maximum plasma concentration (t max), area under the plasma concentration–time curve from hour 0 to the last measurable plasma concentration (AUClast), area under the plasma concentration–time curve extrapolated to infinity (AUC∞), and half-life (t 1/2). These pharmacokinetic metrics permit assessment of the rate and extent of drug absorption (bioavailability). AUC∞ is an index of total drug exposure and C max is an index of maximum or peak exposure to treatment. The AQ, calculated as C max/t max, is a measure of the average rate of rise in concentration between dosing and t max [12]. Because more rapid exposure to higher opioid concentrations correlates with greater drug-liking, higher AQ scores predict greater abuse potential [6, 11, 12].
2.5 Intranasal Tolerability
Pharmacodynamic measurements were assessed by evaluating intranasal tolerability. Subjects rated intranasal discomfort, itching, burning, pain, runny nose, and stuffiness on numeric scales ranging from 0 (none) to 10 (worst I can imagine). Ratings measured only the naris used for drug administration. Intranasal tolerability rating scales were evaluated at predose and at 0.25, 0.5, 0.75, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0, 6.0, 8.0, 10, and 12 h postdose. At approximately 5 and 24 h after each dosing, subjects provided a written response to the question, “What effects (if any) did snorting this drug cause to the inside of your nose?” Additionally, a specialist in ear, nose, and throat procedures performed intranasal endoscopies. These were conducted at predose, and as close to 0.5 h postdose as possible, but not later than 2 h postdose.
2.6 Safety Measures
Safety assessments included reports of adverse events (AEs), clinical laboratory test results, vital signs results, pulse oximetry (SpO2), physical examinations, and electrocardiograms (ECGs). Reported AEs were coded according to the Medical Dictionary for Regulatory Activities (version 9.1) by preferred term and system organ class. AEs reported herein are those that emerged, reemerged, and/or worsened in severity during treatment and were classified as treatment-emergent AEs.
2.7 Statistical Analysis
Subjects who were randomized, received study drug, and had at least one postdose safety assessment were included in safety analyses; subjects who were randomized, received study drug, and had at least one valid pharmacokinetic metric were included in pharmacokinetic analyses. For pharmacokinetic data, a mixed-model analysis of variance was used to compare logarithmic-transformed (base e) values for C max, AUClast, and AUC∞ of oxycodone, with fixed effects for treatment, period, and sequence, as well as random effects of subject within sequence.
For pharmacokinetic data, bioequivalence was indicated if the 90 % confidence intervals (CIs) for oxycodone for a given measure fell entirely within the 80–125 % range. Intranasal tolerability rating scales used descriptive statistics for raw data and for differences from predose for each time point and treatment. The maximum response over time was tabulated and an analysis of covariance model was conducted to assess differences among treatments related to maximum response. This pharmacodynamic model included a covariate for the predose assessment, fixed effects for treatment, period, and sequence, and a random effect of the subject within sequence.
3 Results
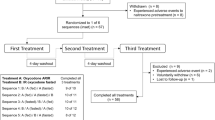
Of 83 subjects screened and enrolled, 30 were randomized (Fig. 1). One was discontinued in period 1 due to the subject’s choice, leaving 29 in treatment periods 2 and 3. The mean (range) age of subjects was 32 (19–52) years. Of the randomized subjects, most were male (66.7 %) and most were white (83.3 %). Mean (range) body weight, height, and body mass index were 78.1 (49.9–94.0) kg, 171.5 (156.0–187.0) cm, and 26.5 (19.1–31.9) kg/m2, respectively.

Study disposition. OC-F finely crushed original formulation of oxycodone-HCl controlled release, ORF-C coarsely crushed reformulated oxycodone-HCl controlled release, ORF-F finely crushed reformulated oxycodone-HCl controlled release
3.1 Pharmacokinetics
After intranasal administration, mean C max was lower for finely crushed ORF and coarsely crushed ORF than that for finely crushed OC (Table 1; Fig. 2). The mean C max metric ratio comparing coarsely crushed ORF and finely crushed OC indicated a 33 % lower C max for coarsely crushed ORF (Table 2). The mean C max metric ratio comparing finely crushed ORF and finely crushed OC indicated a 22 % lower C max for finely crushed ORF. In addition, median t max values were reached more rapidly for finely crushed OC than for either finely crushed ORF or coarsely crushed ORF (Table 1; Fig. 2). The highest mean AQ score was observed for finely crushed OC (31.7 ng/mL per h) compared with either ORF preparation (Fig. 3), with mean AQ scores for finely crushed ORF and coarsely crushed ORF approximately 66 and 80 % lower, respectively, versus finely crushed OC.

Mean plasma oxycodone concentrations following intranasal dosing over time in the full analysis set. OC-F finely crushed original formulation of oxycodone-HCl controlled release, ORF-C coarsely crushed reformulated oxycodone-HCl controlled release, ORF-F finely crushed reformulated oxycodone-HCl controlled release

Abuse quotient box and whisker plots. The open circles represent the individual abuse quotient values for each patient. C max maximum observed plasma concentration, OC-F finely crushed original formulation of oxycodone-HCl controlled release, ORF-C coarsely crushed reformulated oxycodone-HCl controlled release, ORF-F finely crushed reformulated oxycodone-HCl controlled release, SD standard deviation, t max time to maximum plasma concentration
After intranasal administration, mean oxycodone AUC∞ values were 124, 134, and 128 ng·h/mL for finely crushed ORF, coarsely crushed ORF, and finely crushed OC, respectively (Table 1). Mean metric ratios between finely crushed ORF, coarsely crushed ORF, and finely crushed OC are illustrated in Table 2. With mean metric ratios falling within the 80–125 % CI range for all treatment comparisons, these results indicated comparability of treatments for average total oxycodone exposure.
3.2 Intranasal Tolerability
Compared with finely crushed OC, insufflation of finely crushed ORF and coarsely crushed ORF produced significantly higher mean ratings of nasal discomfort (P = 0.0030 and P < 0.0001, respectively) and intranasal stuffiness (P < 0.0001 for each) (Fig. 4). Insufflation of finely crushed ORF produced a significantly lower mean rating for runny nose than finely crushed OC (P = 0.0363), and coarsely crushed ORF yielded significantly greater intranasal discomfort than finely crushed ORF (P = 0.0106). There were no significant differences noted for other measures of nasal tolerability (burning, itching, or pain).

Comparison of maximum response over time on the Intranasal Tolerability Rating Scale (ITRSa) in the randomized safety population. aITRS was rated on an 11-point scale, ranging from 0 (none) to 10 (worst I can imagine). bLS means from ANCOVA. cDifference of means between treatments from ANCOVA. d90 % CI for difference of means from ANCOVA. e P value for pairwise comparison between treatments from ANCOVA. *P < 0.05 between treatment groups. ANCOVA analysis of covariance, CI confidence interval, ITRS Intranasal Tolerability Rating Scale: ITRS was rated on an 11-point scale, ranging from 0 (none) to 10 (worst I can imagine), LS least squares, OC-F finely crushed original formulation of oxycodone-HCl controlled release, ORF-C coarsely crushed reformulated oxycodone-HCl controlled release, ORF-F finely crushed reformulated oxycodone-HCl controlled release
Intranasal endoscopy at 0.5 h after insufflation of either ORF preparation revealed a coarse, white, gel-foam-type material in the nasal passages of subjects, whereas nasal passages were clear after insufflation of finely crushed OC. Subject comments corroborated these findings (see representative data in Fig. 5).

Representative intranasal endoscopy results. OC-F finely crushed original formulation of oxycodone-HCl controlled release, ORF-C coarsely crushed reformulated oxycodone-HCl controlled release, ORF-F finely crushed reformulated oxycodone-HCl controlled release
3.3 Safety
No deaths or serious adverse events were reported. Of the 93 AEs reported, 25 were reported in 15 subjects with finely crushed ORF, 29 were reported in 12 subjects with coarsely crushed ORF, and 39 were reported in 9 subjects with finely crushed OC. One subject chose to discontinue from the study due to an AE of nausea with finely crushed OC (10 mg) dosing. The most commonly reported AEs were headache, nausea, and vomiting and most AEs were of mild or moderate severity. The number and percentage of distinct subjects with AEs that were judged to be related to study treatment are shown in Table 3. All AEs resolved by study end. The eight AEs that were severe and related to treatment were vomiting (3), nausea (3), headache (1), and pain in extremity (1); these were reported by four subjects. Results of laboratory tests, vital signs measurements, SpO2 evaluations, and ECG recordings revealed no clinically significant abnormalities and raised no safety concerns for the study treatments.
4 Discussion
This study compared the effects of original OxyContin® (OC) and reformulated OxyContin® (ORF) on the pharmacokinetic, pharmacodynamic, and safety profiles of healthy men and women. ORF, with its abuse-deterrent properties of reduced crushability showed the intended effects of reduced C max, delayed t max, reduced AQ, and significantly increased intranasal intolerability compared with OC. The safety profile of ORF was similar to that of OC. Most AEs for all treatments were classified as mild or moderate and were those commonly associated with opioid use (e.g., nausea, vomiting, and headache).
Subjects reported greater intranasal discomfort, stuffiness, and intranasal obstruction with ORF than with OC. Decreased subject-reported tolerability and visual demonstrations of increased deposition of undissolved, crushed ORF suggest that recreational abusers, intent on using intranasal administration of prescription opioids, may find that ORF has low desirability. The current study examines acute tolerability of a single-dose administration, so the impact of chronic intranasal abuse of ORF cannot be determined. However, medical complications involving the nasal mucosa with chronic intranasal substance abuse have been documented [13–15].
This study focused on OxyContin® abuse via the intranasal route of administration (i.e., snorting), which is a commonly used method of abuse [7]. OC can be easily crushed to disable its controlled-release properties [6, 8, 16, 17]. The pharmacokinetic profile of orally administered, crushed OC has been shown to be similar to that of immediate-release oxycodone [18]. In addition, intranasal administration of crushed OC resulted in rapid absorption, with most (approximately 75 %) of the administered dose being absorbed, indicating that intranasal administration after crushing is an efficient means of defeating the controlled-release properties of original OxyContin® [8]. This is not unexpected given evidence showing the nasal mucosa is an efficient absorptive surface for agents delivered in powdered form [19, 20]. The rapid rate of absorption that occurs with intranasal nonmedical administration of opioids may increase their abuse potential. Furthermore, recent findings suggest that rate of infusion of opioids, including intravenous morphine [21] and oxycodone [22], is an important determinant of the reinforcing effects of these agents, with rapid infusion resulting in greater effects.
The ultimate goal of the development of ORF was to replace a readily abusable product with an abuse-deterrent product that reduced the frequency and adverse consequences of abuse while maintaining access to treatment for patients with moderate to severe pain who require a continuous, around-the-clock opioid analgesic for an extended period of time. Although no opioid product can ever be abuse-proof [1, 2], abuse-deterrent products offer the possibility of incremental or more substantial reductions in abuse potential compared with previously developed products that are more easily altered, depending upon the product/formulation, the route, the tampering method, and the population [2].
The current study, illustrating the differences in pharmacokinetic profile of intranasal administration of OC and ORF, includes elements that have been recommended for assessing the pharmacokinetic profile of an abuse-deterrent product [3, 23]. Pharmacokinetic and safety analyses are one of four critical components used to characterize the abuse potential of ORF. The other components consist of in vitro tamper-testing studies conducted during formulation development [9], abuse-liking studies [24, 25], and, lastly, epidemiological studies. The real-world impact of any abuse-deterrent product needs to be established by a variety of epidemiologic studies conducted in real-world settings [1–3, 11, 12]. Those epidemiologic studies are currently in progress for reformulated OxyContin® [26]. Recently reported findings appear to support the laboratory-based tamper-testing and abuse potential studies for ORF, including decreased rates of self-reported nonoral abuse in a large national interview survey database [27]; decreased rates of intranasal abuse in a cohort of OxyContin® abusers in rural Kentucky [28]; and decreased incidence of diversion and a decrease in reported street value in surveys from law enforcement agencies [29, 30].
One limitation of the present study is that statistical significance testing using P values was not done. Another limitation is that the endpoints were for pharmacokinetics and safety only, and standardized measures of “drug liking” were not incorporated. The results of the present study can be more clearly interpreted in light of subsequent research looking at both pharmacokinetics and the pharmacodynamics of drug liking.
In January 2013, the US Food and Drug Administration (FDA) released its guidance, Abuse-Deterrence Opioids—Evaluation and Labeling [31]. This guidance proposes that summary statements and label claims regarding abuse deterrence can be based on pharmacokinetic data. The present study employed the major design elements recommended by the FDA in this guidance: the use of the most common route of administration for abuse, the impact of food and alcohol taken into consideration in the design of the study, the measurement of the rate of rise of drug concentration, the inclusion of relevant pharmacokinetic parameters, and the documenting of AEs.
5 Conclusions
In contrast to the original formulation, reformulated OxyContin® administered intranasally demonstrated a lower C max, a longer t max, and a lower AQ when compared with the original formulation. Crushed reformulated OxyContin® was also associated with greater intranasal irritation than crushed original OxyContin®, with otherwise similar safety profiles. Overall, these data suggest that reformulated OxyContin® has a reduced abuse potential compared to the original formulation upon intranasal administration.
References
Schneider JP, Matthews M, Jamison RN. Abuse-deterrent and tamper-resistant opioid formulations: what is their role in addressing prescription opioid abuse? CNS Drugs. 2010;24(10):805–10.
Hamed E, Moe D. Development of tamper deterrent formulations: state of the pharmaceutical industry. Curr Drug Abuse Rev. 2010;3(3):139–46.
Katz NP, Adams EH, Chilcoat H, et al. Challenges in the development of prescription opioid abuse-deterrent formulations. Clin J Pain. 2007;23(8):648–60.
Osgood ED, Eaton TA, Trudeau JJ, et al. A brief survey to characterize oxycodone abuse patterns in adolescents enrolled in two substance abuse recovery high schools. Am J Drug Alcohol Abuse. 2012;38(2):166–70.
Inciardi JA, Surratt HL, Kurtz SP, et al. Mechanisms of prescription drug diversion among drug-involved club- and street-based populations. Pain Med. 2007;8(2):171–83.
Katz N, Dart RC, Bailey E, et al. Tampering with prescription opioids: nature and extent of the problem, health consequences, and solutions. Am J Drug Alcohol Abuse. 2011;37(4):205–17.
Young AM, Havens JR, Leukefeld CG. Route of administration for illicit prescription opioids: a comparison of rural and urban drug users. Harm Reduct J. 2010;7:24.
Lofwall MR, Moody DE, Fang WB, et al. Pharmacokinetics of intranasal crushed OxyContin and intravenous oxycodone in nondependent prescription opioid abusers. J Clin Pharmacol. 2012;52(4):600–6.
Cone EJ, Giordano J, Weingarten B. Laboratory based in vitro tamper testing of reformulated OxyContin: an iterative and incremental scientific approach. Poster presented at the College on Problem of Drug Dependence’s 74th Annual Meeting. June 10–14, 2012; Palm Springs, CA.
Lugo RA, Kern SE. The pharmacokinetics of oxycodone. J Pain Palliat Care Pharmacother. 2004;18(4):17–30.
Webster LR, Bath B, Medve RA. Opioid formulations in development designed to curtail abuse: who is the target? Expert Opin Investig Drugs. 2009;18(3):255–63.
Webster LR. Oxycodone extended-release using gel-cap technology to resist alteration and abuse for the treatment of moderate-to-severe pain. Pain Manage. 2011;1(5):417–25.
Greene D. Total necrosis of the intranasal structures and soft palate as a result of nasal inhalation of crushed OxyContin. Ear Nose Throat J. 2005;84(8):512,514,516.
Yewell J, Haydon R, Archer S, et al. Complications of intranasal prescription narcotic abuse. Ann Otol Rhinol Laryngol. 2002;111(2):174–7.
Jewers WM, Rawal YB, Allen CM, et al. Palatal perforation associated with intranasal prescription narcotic abuse. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99(5):594–7.
Raffa RB, Pergolizzi JV Jr. Opioid formulations designed to resist/deter abuse. Drugs. 2010;70(13):1657–75.
Lipman AG. What have we learned from OxyContin? J Pain Palliat Care Pharmacother. 2003;17(1):1–4.
Webster LR, Bath B, Medve RA, et al. Randomized, double-blind, placebo-controlled study of the abuse potential of different formulations of oral oxycodone. Pain Med. 2012;13(6):790–801.
Ugwoke MI, Agu RU, Verbeke N, et al. Nasal mucoadhesive drug delivery: background, applications, trends and future perspectives. Adv Drug Deliv Rev. 2005;57(11):1640–65.
Matsuyama T, Morita T, Horikiri Y, et al. Influence of fillers in powder formulations containing N-acetyl-l-cysteine on nasal peptide absorption. J Control Release. 2007;120(1–2):88–94.
Marsch LA, Bickel WK, Badger GJ, et al. Effects of infusion rate of intravenously administered morphine on physiological, psychomotor, and self-reported measures in humans. J Pharmacol Exp Ther. 2001;299(3):1056–65.
Comer SD, Ashworth JB, Sullivan MA, et al. Relationship between rate of infusion and reinforcing strength of oxycodone in humans. J Opioid Manag. 2009;5(4):203–12.
US Department of Health and Human Services. Guidance for Industry: Assessment of Abuse Potential of Drugs. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM198650.pdf. Accessed 11 Sept 2012.
Perrino PJ, Colucci S, Harris S, et al. Evaluation of abuse potential of crushed and intranasally administered oxycodone tablets. Abstract presented at the College on Problem of Drug Dependence’s 74th Annual Meeting. June 10–14, 2012; Palm Springs, CA.
Sellers E, Perrino PJ, Harris S, et al. Relative attractiveness of reformulated OxyContin®: comparative assessment of tampering potential and recreational drug user preferences for opioid formulations. Abstract presented at the College on Problem of Drug Dependence’s 74th Annual Meeting. June 10–14, 2012; Palm Springs, CA.
Coplan PM, Chilcoat HD, Baumgartner T, et al. Design of a post-marketing study program to assess the effects of a reformulated extended-release oxycodone tablet on its abuse [abstract]. Pharmacoepidemiol Drug Saf. 2012;21(suppl 3):446 (Abstract 963).
Butler SF, Cassidy TA, Chilcoat H, et al. Abuse rates and routes of administration of reformulated OxyContin: initial findings from a sentinel surveillance sample of individuals assessed for substance abuse treatment. J Pain. 2012. (Published online ahead of print).
DeVeaugh-Geiss A, Leukefeld C, Havens J, et al. Abuse of extended-release (ER) and immediate-release oxycodone in Kentucky following introduction of reformulated ER oxycodone. Abstract presented at: PAIN Week 2012. September 5–8, 2012; Las Vegas, NV.
Davis J, Severtson SG, Bucher-Bartelson B, et al. Reduction in extended release (ER) oxycodone diversion rates following the introduction of a reformulated ER oxycodone product. Abstract presented at: International Association for the Study of Pain’s 14th World Congress on Pain; August 27–21, 2012; Milan, Italy.
Bucher-Bartelson B, Severtson S, Davis J, et al. A comparison of the street price of original and reformulated ER oxycodone. Abstract presented at: International Association for the Study of Pain’s 14th World Congress on Pain; August 27–21, 2012; Milan, Italy.
US Department of Health and Human Services. Guidance for Industry: abuse-deterrent opioids—evaluation and labeling. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM334743.pdf. Accessed 19 March 2013.
Acknowledgments
Role of funding source This study was sponsored by Purdue Pharma L.P. (Stamford, CT, USA). Writing assistance for this publication was funded by Purdue Pharma L.P. and was provided by Jennifer Steeber, PhD, and Huda Abdullah, PhD, formerly of SCI Scientific Communications & Information (Parsippany, NJ, USA). Further writing and editorial assistance was provided by Henry Andrew Caporoso, MA, a full-time employee of Purdue Pharma L.P.
Authors’ contributions
All authors were involved in the design of the study; collection, analysis, and interpretation of data; writing the report; and the decision to submit the manuscript for publication. All authors confirm that this article is an accurate representation of the study results.
Conflict of interest statements/financial disclosure statement
All authors affiliated with Purdue Pharma L.P. are employees. Dr. Apseloff was the primary investigator for this study and provides paid consulting services for Purdue Pharma L.P.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited. The exclusive right to any commercial use of the article is with Springer.
About this article
Cite this article
Perrino, P.J., Colucci, S.V., Apseloff, G. et al. Pharmacokinetics, Tolerability, and Safety of Intranasal Administration of Reformulated OxyContin® Tablets Compared with Original OxyContin® Tablets in Healthy Adults. Clin Drug Investig 33, 441–449 (2013). https://doi.org/10.1007/s40261-013-0085-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-013-0085-x