
Abstract
Background and Objectives
Ezetimibe/simvastatin combination tablet has been approved for the treatment of high low-density lipoprotein cholesterol (LDL-C) levels in patients with primary hypercholesterolaemia or mixed hypercholesterolaemia as adjunctive therapy to diet, when diet alone is insufficient in lowering cholesterol. The aims of this study were to assess the pharmacokinetics and safety of an ezetimibe/simvastatin combination tablet after oral single-dose administration in healthy Chinese subjects including sex-related differences in pharmacokinetics.
Methods
This was an open-label, single-dose study. Twelve healthy subjects (six males and six females) received a single dose of an ezetimibe/simvastatin combination tablet (ezetimibe 10 mg and simvastatin 40 mg). The pharmacokinetic parameters for ezetimibe and simvastatin were assessed by determining total ezetimibe, free ezetimibe, simvastatin and simvastatin acid concentrations using a validated liquid chromatography–tandem mass spectrometry method. Safety was evaluated by monitoring adverse events, laboratory assays, vital signs, physical examinations and 12-lead electrocardiograms.
Results
The pharmacokinetic parameters (mean ± SD) for total ezetimibe and free ezetimibe following a single dose were: maximum plasma drug concentration (Cmax) 81.56 ± 26.62 and 9.40 ± 6.17 ng/mL; time to reach Cmax (tmax) 0.93 ± 0.30 and 1.25 ± 1.27 h; elimination half-life (t½) 24.32 ± 13.27 and 18.90 ± 9.66 h, and mean area under the plasma concentration–time curve from time zero to the time of the last measurable concentration (AUClast) 579.06 ± 241.45 and 126.01 ± 69.01 ng·h/mL, respectively. The pharmacokinetic parameters (mean ± SD) for simvastatin and simvastatin acid following a single dose were: Cmax 11.92 ± 5.50 and 3.37 ± 1.78 ng/mL, tmax 0.98 ± 0.28 and 3.73 ± 1.68 h, t½ 4.19 ± 1.81 and 7.65 ± 7.96 h, and mean AUClast 33.63 ± 20.41 and 32.50 ± 18.79 ng·h/mL. Higher AUClast and AUC from time zero to infinity (AUC∞), and lower apparent total body clearance of drug from plasma after oral administration (CL/F) for total ezetimibe and free ezetimibe were observed in female subjects compared with those in male subjects. There were no differences between the pharmacokinetic parameters of simvastatin and simvastatin acid for female and male subjects in the study.
Conclusion
Ezetimibe/simvastatin combination tablet has a generally favourable safety and tolerability profile in healthy Chinese subjects. A statistically significant difference with regard to sex in the pharmacokinetics of ezetimibe was observed. Sex had no effect on the pharmacokinetics of simvastatin and simvastatin acid.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Simvastatin is a selective 3-hydroxy-3-methyl-glutaryl-coenzyme-A (HMG-CoA) reductase inhibitor [1], and has been widely used for the treatment of hypercholesterolaemia. After oral administration, it is quickly hydrolysed to its active β-hydroxyacid form, simvastatin acid. HMG-CoA reductase is an essential enzyme involved in the synthesis of cholesterol. Consequently, simvastatin reduces low-density lipoprotein cholesterol (LDL-C), triglyceride (TG) and total cholesterol (TC), and increases high-density lipoprotein cholesterol (HDL-C).
Statin therapy remains the mainstay of lipid-lowering management. However, some patients, particularly those at high cardiovascular disease (CVD) risk, do not achieve intensive LDL-C-lowering goals on statin therapy alone [2]. At the highest doses of statin, LDL-C reductions are significant but not well tolerated by all patients [3]. Therefore, in order to achieve greater reductions in LDL-C and minimize increases in the incidence of adverse reactions, it may be necessary to combine statins with an additional drug that reduces LDL-C levels through a different mechanism. Ezetimibe is a selective inhibitor of intestinal cholesterol and related phytosterol absorption that blocks the Niemann-Pick C1-Like 1 protein sterol transporter [4]. Ezetimibe monotherapy produces an approximately 18 % reduction in LDL-C and is well tolerated compared with placebo [5–8]. When ezetimibe is co-administered with simvastatin, significant incremental reductions in LDL-C and TG as well as increases in HDL-C are achieved compared with ezetimibe and statin monotherapies [9–17]. The co-administration of ezetimibe with simvastatin is generally safe and well tolerated [9–18]. Therefore ezetimibe combined with simvastatin can provide greater lipid-altering efficacy than statins alone.
Ezetimibe/simvastatin combination tablet (Vytorin, Merck/Schering-Plough) was approved by the US Food and Drug Administration in July 2004 for the treatment of high LDL-C levels in patients with primary hypercholesterolaemia or mixed hypercholesterolaemia as an adjunctive therapy to diet, when diet alone is insufficient in lowering cholesterol. Ezetimibe/simvastatin combination tablet is bioequivalent to co-administration of ezetimibe and simvastatin as individual tablets [19]. Drug-drug interactions have not been observed when ezetimibe is given concomitantly with simvastatin [20, 21]. Pharmacokinetic data [22, 23] show that ezetimibe is rapidly absorbed and extensively conjugated to a pharmacologically active phenolic glucuronide (ezetimibe-glucuronide) after oral administration. Mean maximum plasma concentrations (Cmax) are generally reached within 1–2 h for ezetimibe-glucuronide and 4–12 h for ezetimibe. After administration of ezetimibe 20 mg, a median maximal plasma concentration (Cmax) of 85 ng/mL is obtained at a median time to reach Cmax (tmax) of 1 h. Ezetimibe is primarily metabolized in the small intestine and liver via glucuronide conjugation with subsequent biliary and renal excretion. Ezetimibe and the active metabolite ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10–20 % and 80–90 % of the total drug in plasma, respectively. The elimination from plasma is slow with an elimination half-life (t½) of about 22 h for both ezetimibe and ezetimibe-glucuronide. Plasma concentration-time profiles exhibit multiple peaks, suggesting enterohepatic recycling. Concomitant food administration (high-fat or non-fat meals) had no effect on the oral bioavailability of ezetimibe when subjects were administered 10-mg tablets. In a multiple-dose study with ezetimibe 10 mg given once daily for 10 days, plasma concentrations for total ezetimibe were slightly higher (<20 %) in women than in men [22]. In another multiple-dose study with ezetimibe 10 mg given once daily for 10 days, plasma concentrations for total ezetimibe were about two-fold higher in older (≥65 years) subjects than those in younger (18–45 years) subjects [22].
Pharmacokinetic data [24] demonstrate that simvastatin, a lactone, is hydrolysed to simvastatin acid after administration. Because of extensive hepatic first-pass extraction, the bioavailability of simvastatin acid following an oral dose of simvastatin is less than 5 % of the dose. Following an oral dose of 14C-labeled simvastatin in man, 13 % of the dose is excreted in urine and 60 % in faeces. Plasma concentrations of total radioactivity (simvastatin plus 14C-metabolites) peak at 4 h and decline rapidly to about 10 % of peak by 12 h post-dose. Food has no effect on the extent of absorption of simvastatin. Although the mean plasma levels for simvastatin and simvastatin acid in the elderly subjects (70–78 years old) were increased by about 45 % compared with younger subjects (18–30 years old), there were no overall differences in safety between older and younger subjects.
The pharmacokinetics and safety of ezetimibe/simvastatin combination tablet (Vytorin) have not been previously studied in healthy Chinese subjects. The present study was designed to (a) evaluate the pharmacokinetics of ezetimibe and simvastatin after a single oral dose of ezetimibe/simvastatin combination tablet (Vytorin) in healthy Chinese subjects; (b) acquire preliminary information about the safety profile of ezetimibe and simvastatin in Chinese subjects; and (c) investigate sex-related differences in the pharmacokinetics of the drugs. The study was conducted in accordance with the regulatory requirements for registration in China.
2 Subjects and Methods
2.1 Study Subjects
Six healthy male and six healthy female subjects between 21 and 24 years of age provided written informed consent and enrolled in the study. Prior to the study, all subjects underwent a physical examination, laboratory testing (including blood chemistry, haematology and urinalysis), 12-lead electrocardiogram and vital signs. Subjects with positive results for HIV, hepatitis B surface antigen and hepatitis C virus were not eligible. Neither prior medications nor concomitant medications during the course of the study were allowed, except for the treatment of adverse events.
The study was conducted in compliance with Good Clinical Practice guidelines [25] and the ethical principles of the Declaration of Helsinki [26]. The study protocol (see Online Resource 1) and informed consent form were approved by the independent ethics committee of ZhongShan Hospital. All subjects were informed by a clinical investigator of the study’s aims and risks, and each subject submitted written informed consent before participating in the study.
2.2 Study Design
This study was an open-label, single-dose pharmacokinetic study in healthy adult male and female subjects. The ezetimibe/simvastatin combination tablet was available as a ezetimibe 10 mg/simvastatin 40 mg tablet which was prepared by Schering-Plough (China) Ltd (manufactured in Singapore by MSD Technology Singapore Pte Ltd). In this study, after an overnight fast of 10 h, each subject received a single oral dose of ezetimibe/simvastatin combination tablet, taken with 180 mL of water. No food was allowed for 4 h after dosing. Additional water intake was permitted 2 h after dosing. Alcoholic beverages, intense physical activity and smoking were not allowed during the study. In the study, all subjects received the same standard weight-maintenance diet that consisted of rice (150 g), fish (200 g), vegetables (250 g), tomato soup (100 g) and oil (15 g) [780 kcal total].
Blood samples of 10 mL for assessment of drug levels were collected in vacutainers containing anticoagulant (heparin) at pre-dose and at 0.5, 0.75, 1.0, 1.25, 1.5, 2.0, 3.0, 4.0, 5.0, 6.0, 8.0, 10.0, 12.0, 16.0, 24.0, 36.0, 48.0, 72.0 and 96.0 h after oral administration of the ezetimibe/simvastatin combination tablet. Blood samples were immediately centrifuged at 1500 g for 10 min at 4 °C to separate the plasma. The separated plasma samples were transferred to a polypropylene tube and stored in the study centre at −20 °C until shipped to the central laboratory where the analyses of ezetimibe and simvastatin concentrations were performed.
2.3 Blood Sample Preparation and Analysis
Plasma samples were analysed by liquid chromatography–tandem mass spectrometry (LC-MS/MS), which was validated in terms of specificity, sensitivity, linearity, within-batch and between-batch precision and accuracy test. This analytical method had some improvements based on other published methods [27, 28] for the determination of ezetimibe and simvastatin.
For the analysis of total ezetimibe (conjugated and unconjugated [free] ezetimibe), 50 μL of plasma sample and 5 μL of internal standard (13C6-ezetimibe, 250 ng/mL) solution were transferred to a 2-mL polypropylene test tube, then 250 μL of sodium acetate buffer (0.5 mol/L, pH 5.0) and 12.5 μL of β-glucuronidase (134,300 IU/mL) were added into the tube. After vortexing for 5 min, the tube was incubated at 50 °C for 60 min and 125 μL of sodium borate solution (0.1 mol/L) was added into it. The mixture was extracted with 1 mL of methyl tert-butyl ether for 10 min by vortexing and then centrifuged at 13,000 rpm for 2 min. The supernatant was transferred to a clean polypropylene tube and dried with a stream of nitrogen gas at 50 °C. The residue was reconstituted with 50 μL of methanol (70 %) and the 10-μL volume was injected into the LC-MS/MS system.
For the analysis of free (unconjugated) ezetimibe, simvastatin and simvastatin acid, 200 μL of plasma sample and 10 μL of internal standard (13C6-ezetimibe 10 ng/mL, D6-simvastatin 25 ng/mL and D6-simvastatin acid 25 ng/mL) solution were transferred to a 2-mL polypropylene test tube. After 10 μL of sodium acetate buffer (0.1 mol/L, pH 4.5) and 1 mL of methyl tert-butyl ether were added, the tube was vortexed for 5 min and then centrifuged at 13,000 rpm for 2 min. The supernatant was transferred to a clean polypropylene tube and dried with a stream of nitrogen gas at 50 °C. The residue was reconstituted with 70 μL of methanol (70 %) and 20-μL volume was injected into the LC-MS/MS system.
Total ezetimibe was detected using negative ionization by multiple reaction monitoring mode under a Sciex API 3000 LC-MS/MS system. The typical ion source parameters were: declustering potential (DP) −40 V, focusing potential (FP) −175 V, entrance potential (EP) −10 V, collision cell exit potential (CXP) −9 V, collision energy (CE) −23 V, and source temperature 450 °C. The mass transitions of m/z 408.2→271.0 and 414.2→271.0 were used to quantify total ezetimibe and 13C6-ezetimibe, respectively. The analytical column was XTerra MS C18 column (50 × 2.1 mm, 3.5 μm). The mobile phase consisted of acetonitrile (A) and 5 mmol/L ammonium acetate (B) [A:B = 55:45, v/v] at a flow rate of 0.25 mL/min.
Free ezetimibe, simvastatin and simvastatin acid were analysed by using negative ionization (within 0–2.35 min) and positive ionization (within 2.35–6 min) by multiple reactions monitoring mode under a Sciex API 3000 LC-MS/MS system. The typical ion source parameters for free ezetimibe, 13C6-ezetimibe, simvastatin, D6-simvastatin, simvastatin acid and D6-simvastatin acid were: declustering potential (DP) −40, −40, 32, 32, −40, −40 V, focusing potential (FP) −175, −175, 140, 140, −220, −220 V, entrance potential (EP) −10, −10, 14, 14, −10, −10 V, collision cell exit potential (CXP) −9, −9, 15, 5, −8, −8 V, collision energy (CE) −23, −23, 17, 17, −24, −24 V, and source temperature 450 °C. The analytical column was XTerra MS C18 column (50 × 2.1 mm, 3.5 μm). The m/z for free ezetimibe, 13C6-ezetimibe, simvastatin, D6-simvastatin, simvastatin acid and D6-simvastatin acid were 408.2→271.0, 408.2→271.0, 419.3→285.3, 425.3→285.3, 435.3→319.1 and 441.3→319.1, respectively. The mobile phase also was composed of acetonitrile (A) and 5 mmol/L ammonium acetate (B) [A:B = 55:45, v/v], but binary gradient elution with a flow rate of 0.25 mL/min was used. The time programme was as follows: 0–0.5 min, 45–15 % B; 0.5–1 min, 15 % B; 1.1–6 min, 45 % B.
The standard curve ranged from 0.25 to 250 ng/mL for total ezetimibe, from 0.02 to 20 ng/mL for free ezetimibe, and from 0.05 to 50 ng/mL for both simvastatin and simvastatin acid. The %RSD (relative standard deviation) values for within- and between-batch precision were <7.1 %, <8.9 %, <7.6 %, and <7.1 % for total ezetimibe, free ezetimibe, simvastatin and simvastatin acid, respectively. Accuracy expressed as bias ranged from −2.43 % to +8.14 %, −2.67 % to +9.71 %, −2.38 % to +3.23 %, and −2.35 % to +2.38 % for total ezetimibe, free ezetimibe, simvastatin and simvastatin acid, respectively. The lower limit of quantification (LLOQ) was 0.25 ng/mL for total ezetimibe, 0.02 ng/mL for free ezetimibe, 0.05 ng/mL for simvastatin and 0.05 ng/mL for simvastatin acid, respectively (signal to noise ratio ≥10). The %RSD values for within-batch precision of LLOQ were 4.22 %, 3.60 %, 7.92 % and 10.18 % for total ezetimibe, free ezetimibe, simvastatin and simvastatin acid, respectively.
2.4 Pharmacokinetic and Statistical Analysis
Non-compartmental pharmacokinetic analysis was used to analyse plasma drug concentration-time data. The elimination rate constant (ke ≈ λz) was determined by linear regression of the logarithm of the concentration in plasma with time over the terminal phase. t½ was calculated as 0.693/ke. Cmax and tmax were read from the observed values. Area under the plasma concentration–time curve from time zero to infinity (AUC∞) was determined by summing the areas from time zero to the time of last quantifiable concentration (AUClast) by trapezoidal and log-trapezoidal methods with the extrapolated area. The extrapolated area was determined by dividing the last detectable concentration by the slope of the terminal log-linear phase. The apparent total clearance (CL/F) was calculated by using the equation CL/F = dose/AUC∞. The apparent volume of distribution (Vd/F) was determined by using the equation Vd/F = CL/F/ke.
Pharmacokinetic parameters were summarized over the set of evaluable subjects by means and SD. The variances of pharmacokinetic parameters between male and female subjects were compared by analysis of variance (ANOVA) for AUC, Cmax, t½, CL/F, Vd/F and non-parametric test for tmax. All analyses of pharmacokinetic data were performed using SPSS version 16.0 (SPSS Inc., Chicago, IL, USA) software. A p-value <0.05 was considered statistically significant.
2.5 Safety
Adverse events, clinical laboratory tests (e.g., haematology—including haematocrit, haemoglobin, reticulocytes, platelets and other measures, clinical chemistries and urinalysis), vital signs, physical examinations and 12-lead electrocardiogram were performed for each subject at screening and at the follow-up period to assess the safety and tolerance of the ezetimibe/simvastatin combination tablet.
3 Results
3.1 Study Characteristics
Six healthy male subjects and six healthy female subjects were included. The demographic details were as follows. The mean (SD) values for males were: age 23.00 (0.89) years (range 22–24 years), weight 66.00 (5.93) kg (range 60–72 kg), height 172.50 (5.24) cm (range 165–181 cm) and body mass index 22.16 (1.34) kg/m2 (range 20.72–23.78 kg/m2). The mean (SD) values for females were: age 22.50 (0.84) years (range 21–23 years), weight 51.83 (4.17) kg (range 46–57 kg), height 161.67 (5.72) cm (range 155–169 cm) and body mass index 19.87 (1.94) kg/m2 (range 18.29–23.73 kg/m2).
3.2 Pharmacokinetic Analysis
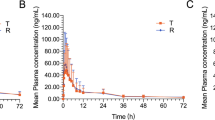
Pharmacokinetic parameters of subject 4 (male) were excluded from the pharmacokinetic analysis because the elimination t½ of 160.99 h and 125.91 h for free ezetimibe and simvastatin acid, respectively, were obviously abnormal, being ten times more than the mean t½ values of the other 11 subjects. The mean concentrations of total ezetimibe, free ezetimibe, simvastatin and simvastatin acid from the plasma-versus-time profiles obtained after administration of a single dose of ezetimibe/simvastatin combination tablet to 11 Chinese subjects are shown in Fig. 1. The results of the pharmacokinetic analysis are summarized in Table 1. The values of the pharmacokinetic parameters are presented as mean ± SD. After oral administration of an ezetimibe/simvastatin combination tablet, a Cmax value of 81.56 ± 26.62 ng/mL of total ezetimibe was rapidly achieved within 0.75–1.5 h. Free ezetimibe slowly appeared in plasma, with a Cmax value of 9.40 ± 6.17 ng/mL occurring within 0.75–5.0 h after administration. The mean AUClast values for total ezetimibe and free ezetimibe were 579.06 ± 241.45 ng·h/mL and 126.01 ± 69.01 ng·h/mL, respectively; the mean AUC∞ values for total ezetimibe and free ezetimibe were 628.65 ± 295.04 ng·h/mL, and 134.02 ± 77.42 ng·h/mL, respectively. Both total ezetimibe and free ezetimibe were eliminated slowly with an elimination t½ of 24.32 ± 13.27 h and 18.90 ± 9.66 h, respectively. Following a single dose of an ezetimibe/simvastatin combination tablet, simvastatin was rapidly absorbed and a Cmax value of 11.92 ± 5.50 ng/mL appeared within 0.75–1.5 h after oral administration. The Cmax of simvastatin acid was 3.37 ± 1.78 ng/mL and was achieved within 0.75–6 h. The mean AUClast values for simvastatin and simvastatin acid were 33.63 ± 20.41 ng·h/mL and 32.05 ± 18.79 ng·h/mL; the mean AUC∞ values for simvastatin and simvastatin acid were 34.28 ± 20.41 ng·h/mL, and 33.76 ± 19.39 ng·h/mL, respectively. Both simvastatin and simvastatin acid were eliminated rapidly with an elimination t½ of 4.19 ± 1.81 h and 7.65 ± 7.96 h, respectively.

Mean plasma versus time concentrations of (a) simvastatin and simvastatin acid, and (b) total ezetimibe and free ezetimibe following a single dose of ezetimibe/simvastatin combination tablet to 11 healthy Chinese subjects
The pharmacokinetic parameters and statistical results between five male (subject 4 was excluded) and six female subjects after a single dose of ezetimibe/simvastatin combination tablet are shown in Tables 2 and 3. In the study, significantly higher AUClast and AUC∞, and lower CL/F of total ezetimibe and free ezetimibe were observed in female subjects compared with males (all differences p <0.05). Other pharmacokinetic parameters of ezetimibe, simvastatin and simvastatin acid showed no statistical difference (p >0.05) between males and females.
3.3 Safety
Ezetimibe/simvastatin combination tablet has a favourable safety and tolerability profile. At follow-up, abnormal increases in creatine kinase values were observed in five subjects. In one subject an increase in creatine kinase was >2x the upper limit of the normal value (ULN), and in the other four subjects increases in creatine kinase levels were mild (<2x ULN). One week after follow-up, creatine kinase values all returned to normal in these five subjects. These reversible increases in creatine kinase were considered to be drug related by the study investigator. In addition, at follow-up one subject’s 12-lead electrocardiogram was reported as being abnormal (second-degree atrioventricular block) and this subject was hospitalized for observation. This adverse event was not considered to be drug related by the study investigator. No clinically significant changes in vital signs and physical examinations and 12-lead electrocardiogram were observed in the other subjects.
4 Discussion
Compared with the known pharmacokinetic data of ezetimibe [22, 23], the pharmacokinetic characteristics of ezetimibe in Chinese healthy subjects were generally consistent with those in non-Chinese subjects. Ezetimibe was rapidly absorbed and conjugated as ezetimibe-glucuronide after oral administration. Both of them were eliminated slowly with a long t½ of approximately 18–24 h. Although the plasma concentration of total ezetimibe was determined in the study, it was equal to the plasma concentrations of ezetimibe-glucuronide and free ezetimibe combined. Therefore, the pharmacokinetic results of this study showed that ezetimibe-glucuronide and ezetimibe constituted about 80–90 % and 10–20 %, respectively, of total drug in plasma, similar to metabolism rates reported previously [23]. Meanwhile, plasma concentration-time profiles of total ezetimibe and free ezetimibe also showed multiple peaks that confirmed the enterohepatic recycling of ezetimibe in vivo.
The current study did suggest the existence of a sex disparity in total ezetimibe and free ezetimibe pharmacokinetics, with two-fold differences in AUC, Cmax and CL/F being observed between males and females. As we know, the effect of sex on the pharmacokinetics may be due to the subject’s sex or to differences in subject weight or renal function between males and females. Therefore, we evaluated whether differences in subject weight or renal function between males and females had an effect on the pharmacokinetics or not in the study. No difference in creatine clearance rate was found between the sexes. After we corrected the pharmacokinetic parameters for subject’s weight, sex-related differences in total ezetimibe and free ezetimibe pharmacokinetics still existed. Therefore in this study, the effect of sex on the pharmacokinetics of ezetimibe is actually due to the subject’s sex, not due to differences in patient weight or renal function between the sexes. A previous study showed the sex-related differences in the pharmacokinetics of ezetimibe had no effect on the clinical therapeutic efficacy and safety of ezetimibe between males and females [29] with no dosage adjustment of ezetimibe/simvastatin necessary on the basis of patient sex [30]. However, in this study compared with previously reported results, these pharmacokinetic differences were more apparent between males and females. Therefore, further clinical studies in China are needed to investigate whether these sex-related pharmacokinetic differences affect the clinical therapeutic efficacy and long-term safety of ezetimibe in males compared with females.
Our study showed that both simvastatin and simvastatin acid were absorbed and eliminated rapidly. However, in our study, AUC and Cmax were higher and t½ was longer than results reported in 24 healthy Caucasian subjects [31]. These differences may be attributed to several reasons. Firstly, the methods for determining simvastatin and simvastatin acid were different. In particular, the sensitivity of the method used in our study was higher than that used in the Caucasian study (0.05 ng/mL and 0.1 ng/mL respectively). Secondly, the blood sample collection times were different. The last blood sample collection time in our study was 96 h, whereas the last collection time in the Caucasian study was 24 h. However, plasma concentrations of simvastatin and simvastatin acid in most subjects were still detectable up to 36 h. Therefore, the inadequate time may have resulted in the low AUC, which represents the low extent of absorption. Thirdly, AUC and Cmax might have been influenced by individual variance and race variance. However, since the number of subjects in our study was small, further clinical studies are needed to evaluate the influence of race on the pharmacokinetics of simvastatin and simvastatin acid. In our study, the pharmacokinetic profiles of simvastatin and simvastatin acid were similar between males and females and no statistical significance was found, which suggested there were no effects of sex on the absorption and elimination of simvastatin and simvastatin acid.
5 Conclusion
The ezetimibe/simvastatin combination tablet has a safety and tolerability profile in healthy Chinese subjects after single oral administration that is similar to that reported previously in other populations. The pharmacokinetics of ezetimibe were consistent with previously published data. A sex-related difference in the pharmacokinetics of ezetimibe was observed in this study. The pharmacokinetics of simvastatin and simvastatin acid in Chinese subjects were different from those reported previously in Caucasian subjects, and a sex-related difference in the pharmacokinetics of simvastatin and simvastatin acid was not found in the study.
References
Isaacsohn J, Hunninghake D, Schrott H, et al. Effects of simvastatin, an HMG-CoA reductase inhibitor, in patients with hypertriglyceridemia. Clinl Cardiol. 2003;26(1):18–24.
Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation. 2009;120(1):28–34.
Gotto AM. Statins, cardiovascular disease, and drug safety. Am J Cardiol. 2006;97(suppl):S3–5.
Altmann SW, Davis HR, Zhu LJ, et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303(5661):1201–4.
Jeu LA, Cheng WM. Pharmacology and therapeutics of ezetimibe (SCH58235), a cholesterol-absorption inhibitor. Clin Ther. 2003;25(9):2352–87.
Bays HE, Moore PB, Drehobl MA, et al. Effectiveness and tolerability of ezetimibe in patients with primary hypercholesterolemia: pooled analysis of two phase II studies. Clin Ther. 2001;23(8):1209–30.
Davis HR. Ezetimibe: first in a new class of cholesterol absorption inhibitors. Int Congr. 2004;1262(5):243–6.
Dujovne CA, Ettiinger MP, McNeer JF, et al. Efficacy and safety of a potent new selective cholesterol absorption, ezetimibe, in patients with primary hypercholesterolemia. Am J Cardiol. 2002;90(10):1092–7.
Bays HE, Ose L, Fraser N, et al. A multicenter, randomized, double-blind, placebo-controlled, factorial design study to evaluate the lipid-altering efficacy and safety profile of the Ezetimibe/simvastatin tablet compared with ezetimibe and simvastatin monotherapy in patients with primary hypercholesterolemia. Clin Ther. 2004;26(11):1758–73.
Anouk G, Cynthia CJ, Maud NV, et al. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;52(17):1421–9.
Nicola A, Alberico LC, Christie MB, et al. Effect of ezetimibe/simvastatin versus atorvastatin or rosuvastatin on modifying lipid profiles in patients with diabetes, metabolic syndrome, or neither: results of two subgroup analyses. J Clin Lipidol. 2008;2(2):91–105.
John RG, Ronald BG, Theodore M, et al. Lipoprotein and apolipoprotein ratios in the VYTAL trial of ezetimibe/simvastatin compared with atorvastatin in type 2 diabetes. J Clin Lipidol. 2008;2(1):19–24.
Davidson MH, McGarry T, Bettis R, et al. Ezetimibe coadministered with simvastatin in patients with primary hypercholesterolemia. J Am Coll Cardiol. 2002;40(12):2125–34.
Pearson T, Ballantyne C, Sisk C, et al. Comparison of effects of ezetimibe/simvastatin versus simvastatin versus atorvastatin in reducing C-reactive protein and low-density lipoprotein cholesterol levels. Am J Cardiol. 2007;99(12):1706–13.
Ballantyne CM, Blazing MA, King TR, et al. Efficacy and safety of ezetimibe co-administered with simvastatin compared with atorvastatin in adults with hypercholesterolemia. Am J Cardiol. 2004;93(12):1487–94.
Robinson JG, Ballantyne CM, Grundy SM, et al. Lipid-altering efficacy and safety of ezetimibe/simvastatin versus atorvastatin in patients with hypercholesterolemia and the metabolic syndrome (from the VYMET Study). Am J Cardiol. 2009;103(12):1694–702.
Ballantyne CM, Abate N, Yuan Z, et al. Dose-comparison study of the combination of ezetimibe and simvastatin (Vytorin) versus atorvastatin in patients with hypercholesterolemia: The Vytorin Versus atorvastatin (VYVA) Study. Am Heart J. 2005;149(3):464–73.
Kashani A, Sallam T, Bheemreddy S, et al. Review of side-effect profile of combination ezetimibe and statin therapy in randomized clinical trials. Am J Cardiol. 2008;101(11):1606–13.
Migoya EM, Bergman A, Hreniuk D, et al. Bioequivalence of an ezetimibe/simvastatin combination tablet and coadministration of ezetimibe and simvastatin as separate tablets in healthy subjects. Int J Clin Pharmacol Ther. 2006;44(2):83–92.
Ballantyne CM. Ezetimibe: efficacy and safety in clinical trials. Eur Heart J 2002;4 (suppl J):J9–J18.
Kosoglou T, Meyer I, Veltri EP, et al. Pharmacodynamic interaction between the new selective cholesterol absorption inhibitor ezetimibe and simvastatin. Br J Clin Pharmacol. 2002;54(3):309–19.
Zetia® (ezetimibe) drug monograph. http://www.pharmacy.cuhk.edu.hk/ampoule/en/monograph/ezetimibe.pdf. Accessed 2010 Nov 1.
Kosoglou T, Statkevich P, Johnson-Levonas AO, et al. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clin Pharmacokinet. 2005;44(5):467–94.
ZOCOR® (Simvastatin) tablets. Merck & Co., INC. Whitehouse Station, NJ08889, USA.
European Medicines Agency (EMEA) Guideline for Good Clinical Practice ICH Topic E 6 (R1). July 2002, CPMP/ICH/135/95. http://www.emea.europa.eu/pdfs/human/ich/013595en.pdf. Accessed 2009 Oct 28.
World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. http://www.wma.net/en/30publications/10polocies/b3/index.html. Accessed 2009 Oct 28.
Li S, Liu G, Jia J, Li X, Yu C, et al. Liquid chromatography-negative ion electrospray tandem mass spectrometry method for the quantification of ezetimibe in human plasma. J Pharm Biomed Anal. 2006;40(4):987–92.
Barrett B, Huclova J, Dohalsky VB, et al. Validated HPLC-MS/MS method for simultaneous determination of simvastatin and simvastatin hydroxyl acid in human plasma. J Pharm Biomed Anal. 2006;41(2):517–26.
Bennett SK, Huttner RP, Lipka L, et al. Efficacy and safety of ezetimibe coadministered with statins in male and female patients. Obstet Gynecol. 2004;109:966–71. (Abstract).
Zetia [package insert]. North Wales (PA): Merck/Schering-Plough Pharmaceuticals, 2005.
Najib NM, Idkaidek N, Adel A, et al. Pharmacokinetics and bioequivalence evaluation of two simvastatin 40 mg tablets (Simvast & Zocor) in healthy human volunteers. Biopharm Drug Dispos. 2003;24(5):183–9.
Acknowledgements
This study was sponsored by Schering-Plough (China) Ltd, which was involved in the design of the study and in reviewing the manuscript. The authors thank Chen Yu, Gang-Yi Liu and Jing-Ying Jia, Shanghai Xuhui Central Hospital, China, for assistance with the blood sample analysis, and Joanne E. Tomassini, Merck, North Wales, PA, USA, for editorial assistance. The authors have indicated that they have no other conflicts of interest with regard to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chu, NN., Chen, WL., Xu, HR. et al. Pharmacokinetics and Safety of Ezetimibe/Simvastatin Combination Tablet. Clin Drug Investig 32, 791–798 (2012). https://doi.org/10.1007/s40261-012-0013-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-012-0013-5