
Abstract
Interleukin-6 (IL-6) signaling is a critical target in inflammatory pathways. Today, tocilizumab (TCZ) and sarilumab (SAR), two IL-6 receptor-inhibiting monoclonal antibodies, are widely used in the treatment of rheumatoid arthritis (RA), with a favorable efficacy/safety profile. Successful introduction of such agents in the treatment of RA has encouraged the development of other agents targeting different points of the pathway. Sirukumab (SRK), a human anti-IL-6 monoclonal antibody, has been evaluated in clinical trials and showed largely similar clinical efficacy compared with TCZ and other IL-6 pathway-targeting agents. Furthermore, the drug safety profile seemed to reflect the profile of adverse effects and laboratory abnormalities seen in other inhibitors of the IL-6 pathway. However, increased death rates under SRK treatment compared with placebo raised safety concerns, which led to the decision by the FDA to decline the approval of SRK in August 2017. However, during the 18-week true placebo-controlled period, mortality rates were identical in the placebo- and SRK-treated patients. Comparisons after week 18 may be confounded by some factors, and also the ‘crossover’ design resulted in various treatment groups with varying drug exposure periods. The limited placebo exposure relative to SRK exposure makes interpretation of mortality rates difficult. We do not know whether the imbalance in mortality rates seen for SRK is a true safety signal or a result of bias due to the study design. Therefore, further long-term clinical data as well as basic research is needed to allow deeper insight into IL-6 signaling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The complexity of the interleukin (IL)-6 pathway in regulating autoimmune and inflammatory cascades is not yet fully understood |
Compared with IL-6R inhibition, direct IL-6 inhibition may have a higher potency in the treatment of systemic complications of rheumatoid arthritis (RA), such as depression and cardiovascular diseases |
Currently, only the use of IL-6 receptor inhibition in the treatment of RA is established, but it is not possible to make a final judgment upon direct IL-6 inhibition with respect to safety concerns |
It is difficult to interpret the mortality rates and long-term safety of sirukumab, because of the limited placebo exposure time relative to sirukumab |
1 Introduction
Interleukin-6 (IL-6) is an important cytokine both for innate and adaptive immunity [1]. Numerous cell types produce IL-6, including monocytes, T cells, fibroblasts and endothelial cells, especially at inflammation sites [2]. The IL-6 pathway is involved in different inflammatory diseases and could be a potential target in a broad spectrum of indications in diverse disciplines of medicine [3]. Better understanding of this pathway has led to the potential for development of several new treatment modalities affecting different points of the pathway [4]. Today, tocilizumab (TCZ) and sarilumab (SAR), two IL-6 receptor (IL-6R) inhibitors, are approved for use in rheumatoid arthritis (RA), in combination with methotrexate (MTX) as well as monotherapy. TCZ is also indicated for systemic as well as polyarticular juvenile idiopathic arthritis, giant cell arteritis, and a cytokine release syndrome. The IL-6 neutralizing antibody (Ab) siltuximab is approved for multicentric Castleman disease (MCD). Several agents targeting IL-6R, IL-6, or the trans-signaling mechanism of the IL-6 pathway are also in late-stage development phases.
In this article, we describe the role of IL-6 signaling in the pathogenesis of RA, describing the molecular interactions between IL-6, IL-6R, the IL-6 transducer molecule gp130, and subsequent signaling cascades. Based on in vitro and preclinical data, we will then discuss the theoretical differences that could arise from the use of monoclonal antibodies (mAb) that bind to IL-6 versus IL-6R, considering also extra-articular effects as well as potential safety considerations. We will address the known clinical efficacy and safety profile for IL-6R and IL-6 mAbs to elaborate on the potential differences and similarities of both approaches.
2 IL-6 Signaling
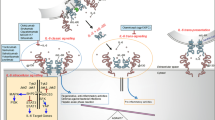
IL-6 was initially recognized as a regulator of the acute-phase response, and for its role in the activation of T cells and differentiation of B cells [5, 6]. It is a pleotropic cytokine and displays hormone-like functions in various situations such as lipid metabolism, insulin resistance, vascular disease, and neuroendocrine regulation [7,8,9]. IL-6 demonstrates its biological activities only by binding to its specific receptor, IL-6R. Neither IL-6 nor IL-6R has a measurable affinity for gp130. However, as a complex, IL-6 and IL-6R can bind to and activate the IL-6R β-subunit, gp130, leading to its dimerization and intracellular signaling (Fig. 1) [2, 10]. The first step in intracellular signaling is the activation of Janus kinases (JAKs), which are constitutively associated with the cytoplasmic tail of gp130. Activation of JAKs by auto-phosphorylation leads to the phosphorylation of the cytoplasmic part of gp130 on five tyrosine residues. The membrane-proximal tyrosine is a docking site for the Src homology 2-containing protein tyrosine phosphatase 2 (SHP2), which activates the mitogen-activated protein kinase (MAPK) and the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3 K) pathways as two signal transducer and activator of transcription (STAT) independent pathways activated by IL-6. Phosphorylation of the remaining four tyrosine residues activates STAT3 by phosphorylation and dimerization and to a lesser extent STAT1. Subsequently, STATs induce the transcription of target genes in the nucleus. Besides the JAK/STAT pathway, IL-6 signaling also stimulates Src-family kinase (SFK)-dependent signaling, which probably leads to the activation of different transcriptional regulators including YES-associated protein (YAP) [11]. Of note, phosphorylation of the tyrosine residue 759 (Tyr759) in the cytoplasmic tail of gp130 is important for negative regulation of IL-6 signal transduction. SHP2 and suppressor of cytokine signaling 3 (SOCS3) bind to this phosphotyrosine and attenuate the IL-6 downstream JAK/STAT signaling [12].

IL-6 signaling cascade. IL-6 demonstrates its biological activities only by binding to its specific receptor, IL-6R. This cytokine-receptor complex then associates with the IL-6R β-subunit, gp130, leading to its dimerization and intracellular signaling. Classical IL-6 receptor signaling occurs in cells that express IL-6R and gp130. IL-6R can be proteolytically cleaved from the cell membrane by ADAM17, generating sIL-6R. This mechanism of trans-signaling allows IL-6 to act on cells that lack IL-6R. Both modes of IL-6 receptor signaling lead to gp130 activation of Janus kinases 1 and 2 and Tyrosine kinase 2, and a series of proximal tyrosine residues that activate STAT1, STAT3, MAPK and PI3K cascade. Besides the JAK/STAT pathway, IL-6 signaling also stimulates SFK-dependent signaling, which probably leads to the activation of different transcriptional regulators including YAP. Phosphorylation of the tyrosine residue 759 in the cytoplasmic tail of gp130 is important for negative regulation of IL-6 signal transduction. SHP2 and SOCS3 bind to this phosphotyrosine and attenuate the IL-6 downstream JAK/STAT signaling. ADAM17 a disintegrin and metallopeptidase domain 17, IL-6 interleukin-6, IL-6R interleukin-6 receptor, Jak Janus kinases, MAPK mitogen-activated protein kinase, mIL-6R membrane bound IL-6R, PI3K phosphatidylinositol-4,5-bisphosphate 3-kinase, SFK Src-family kinase, SHP2 Src homology 2-containing protein tyrosine phosphatase 2, sIL-6R soluble IL-6R, SOCS3 suppressor of cytokine signaling 3, STAT signal transducer and activator of transcription, Tyk2 Tyrosine kinase 2, Tyr759 tyrosine residue 759, YAP YES-associated protein
IL-6 has no binding affinity to gp130 in the absence of IL-6R [13]. Remarkably, while all cells of the body express gp130, the surface expression of the IL-6R is restricted to hepatocytes, leukocytes, and some epithelial cells [2, 11].
However, the biological activity of IL-6 is not restricted to the aforementioned cells, since IL-6R can be proteolytically cleaved from the cell membrane by a disintegrin and metallopeptidase domain 17 (ADAM17), generating a soluble IL-6R (sIL-6R) [14]. This soluble form of IL-6R in combination with gp130 can also bind IL-6 in order to initiate intracellular signaling, which is called ‘trans-signaling.’ The signaling of IL-6 via membrane-bound IL-6R (mIL-6R) is called ‘classic signaling.’ Heink et al. have also described a novel mechanism of IL-6 signaling, called ‘trans-presentation’, in which T cells respond to IL-6 in the absence of IL-6Rα expression [15]. Membrane-bound IL-6Rα in complex with IL-6 is presented by dendritic cells and sensed by gp130 molecules expressed on T cells. Trans-presentation of IL-6 by dendritic cells is required for priming of pathogenic T-helper 17 (TH17) cells. The authors reported that, while similar to classic IL-6 signaling and IL-6 trans-signaling, trans-presentation remains amenable to neutralization via anti-IL-6Rα antibodies. Anti-IL-6 antibodies fail to inhibit IL-6 trans-presentation. This finding is further evidence of the complexity of the IL-6 pathway, which necessitates careful evaluation during development of novel therapeutic strategies. However, these observations shown in mice need to be further evaluated in human cells.
3 Soluble gp130
A soluble form of gp130 (sgp130) is naturally found in the circulation and together with sIL-6R it is thought to buffer circulating IL-6 levels [16, 17]. Since the capacity of the buffer increases with the increased levels of sIL-6R, the interaction of both molecules, sgp130 and sIL-6R, inhibits the stimulation of gp130-expressing cells and represents a protective barrier against inflammatory diseases [18]. Of note, carriers of a human IL6R gene (rs7529229) polymorphism, which causes an Asp358Ala substitution in the ADAM17 cleavage site of the IL-6R protein, have increased levels of sIL-6R in the circulation and are protected from various autoimmune diseases including RA [19, 20]. The levels of sIL-6R in the human circulation have been described to be in the range of 50–75 ng/mL [11]. However, it is not clear whether these levels can predict the required amount of an IL-6R blocking agent for clinical trials and whether the amount of IL-6R blocking agent affects the relative blocking of sIL-6R compared with mIL-6R.
The design of a fusion protein of the soluble extracellular portion of gp130 with the constant portion of the human immunoglobulin (Ig) G1 antibody led to new insights into the IL-6 pathway. This molecule, termed sgp130Fc, was shown to exclusively block IL-6 trans-signaling without affecting classic signaling both in human and animals [21]. Inhibition of trans-signaling with sgp130Fc was shown to be effective in mouse models of inflammatory bowel disease, RA, peritonitis, asthma, and colon cancer [22,23,24,25,26,27,28,29]. Moreover, this blockade did not compromise the immune response to infection in experimental models of listeria infection and tuberculosis [30, 31]. Therefore, these data suggest that pro-inflammatory effects of IL-6 occur via the trans-signaling pathway, while anti-inflammatory activities, regenerative functions, and protection against infections occur via classic signaling (Fig. 1) [3]. The sgp130Fc protein (olamkicept) has successfully passed phase I clinical trials and a phase II trial is currently underway in patients with inflammatory bowel disease [32] (ClinicalTrials.com identifier NCT03235752). However, the product is not currently being evaluated in RA.
4 IL-6 and Rheumatoid Arthritis
In patients with RA, elevated serum levels of both IL-6 and IL-6R are found in serum and synovial fluid of affected joints [33,34,35]. Moreover, IL-6 levels correlate with surrogate markers of disease activity such as rheumatoid factor, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) [36]. IL-6 levels have also been found to correlate with disease manifestations, including the number of inflamed joints and early morning stiffness. Patients treated with disease-modifying antirheumatic drugs (DMARDs) show decreases in IL-6 levels, which correlate with improvements in the number of inflamed joints and morning stiffness [37]. In animal models of RA, while mice bearing a gp130 mutation (which causes excess IL-6 signaling) develop RA-like joint disease, IL-6 deficient mice are resistant to development of collagen-induced or chronic autoimmune arthritis [38,39,40]. Finally, the successful introduction of TCZ into the treatment of RA has established the role of IL-6 signaling in patients with RA.
IL-6 is important for coordination of the activity of the innate and adaptive immune systems [41]. It regulates leukocyte and T-cell differentiation, proliferation, trafficking, survival (control of apoptotic and anti-apoptotic factors), antibody production, and specificity (class switching, somatic hypermutation). IL-6 has a pivotal role in the differentiation of T-helper (TH) cells and TH17 cells, which are important in inflammatory states including RA [42, 43]. In mice, transforming growth factor (TGF)-β induces the differentiation of regulatory T (TREG) cells, which are known to inhibit T-cell activation. However, the combination of IL-6 and TGF-β induces TH17 differentiation [42]. In human T cells, it was shown that sIL-6R in combination with IL-6 was more effective in the stimulation of TH17 cells than IL-6 alone [44, 45]. Moreover, the anti-IL-6R antibody TCZ increases CD39+ TREG cells in mice models of arthritis as well as in patients with RA; however, TH17 cells are not affected [46].
5 IL-6 Inhibition Versus IL-6 Receptor Inhibition
The rationale for developing IL-6 inhibitors includes the fact that there is less circulating cytokine versus receptor (100- to 1000-fold less), which was supported by linear pharmacokinetics, and lower drug load compared with the IL-6 receptor antagonist observed in sirukumab (SRK) studies [34, 47]. In contrast to steady-state mean serum SRK trough concentrations (Ctrough) of 0.99–11.63 μg/mL for the SRK treatment groups in Part B of the study by Smolen et al., steady state Ctrough TCZ concentrations were 5.9 ± 6.3, 18.7 ± 15.3, and 45.3 ± 22.2 μg/mL for 162 mg subcutaneous (SC) every 2 weeks, 8 mg/kg intravenous (IV) every 4 weeks, and 162 mg SC weekly doses from the SUMMACTA and BREVACTA study data [48,49,50,51]. Another rationale for developing IL-6 inhibitors is the presence of IL-6 receptor gene polymorphisms, which was also supported by the observation that variations in single nucleotide polymorphisms known to impact the efficacy of TCZ do not impact the efficacy of SRK [47, 52].
Up to now, blockade of IL-6 and IL-6R generally displayed similar effects; however, translation of basic knowledge regarding the molecular mechanisms of the differences between these two strategies reminds us about possible differences we might face in terms of clinical situations. For example, the human IL-6R serves also as the receptor for ciliary neurotrophic factor (CNTF) and IL-30 (p28) [53, 54]. The binding of human CNTF to the respective IL-6R occurs with an affinity roughly 50-fold lower compared with IL-6 [53]. CNTF is a neurotrophic cytokine that exerts neuroprotective effects in multiple sclerosis and amyotrophic lateral sclerosis. Clinical application of human CNTF, however, was prevented by high toxicity at higher dosages [55]. The clinical side effects observed were cachexia, aseptic meningitis, respiratory failure, and reactivation of viral infections. It may be hypothesized that inhibition of IL-6 may lead to overstimulation of IL-6R by CNTF and potentially associated adverse effects (AEs). However, functions of IL-30 have yet to be elucidated in human cells. IL-30 is a cytokine subunit of IL-27. Studies up to now showed both proinflammatory and anti-inflammatroy effects in human cells [56, 57]. Petes et al. reported inhibition of IL-27- and IL-30–mediated inflammatory responses by IL-6 in human monocytes [58]. They also demonstrated a role for sIL-6Rα and gp130 in IL-30–mediated activity in human cells.
In contrast to T cells, only a small percentage of B cells express IL-6Rα [30]. Zhang et al. demonstrated that in the absence of IL-6Rα, IL-6 can induce STAT3 activation by binding to CD5 in CD5+ B cells [59]. They concluded that their results raised the possibility that other cell types may also use CD5 to respond to IL-6, thereby contributing to STAT3 activation.
Another interesting study showed differences with respect to IL-6 versus IL-6R deficiency using a model of IL-6Rα-deficient mice [60]. These animals share inflammatory deficits seen in IL-6-deficient mice, but they did not display a delay in wound healing. As a surprising finding, mice with a combined deficit of IL-6 and IL-6Rα, or IL-6-deficient mice treated with an IL-6Rα–blocking Ab, demonstrated improved wound healing relative to mice with sole IL-6 deficiency. Thus, inhibition of only IL-6 without inhibiting IL-6R interferes with proper wound healing and may be associated with morbidity and mortality in IL-6 blockage. These findings indicate that the IL-6 pathway may have a more complicated mechanism than we suppose in regulating autoimmune and inflammatory cascades.
Preliminary data also suggest a beneficial role of targeting IL-6 cytokine for systemic complications associated with RA, such as depression and cardiovascular disease (CVD) [61]. Significant elevation of IL-6 levels in the plasma of patients with depression, together with the shown higher prevalence of depression in patients with RA, led to evaluation of the efficacy of the IL-6 pathway targeting agents on depressive symptoms of RA patients [62, 63]. In a post-hoc analysis of the randomized phase II study, SRK significantly improved the depressed mood and anhedonia symptoms of the RA patients independently of clinical response [48, 64]. Of note, 25% of patients had these symptoms and the response to therapy was significantly associated with the baseline sIL-6R levels. In an analysis of 11 clinical and preclinical studies, SRK seems to be a promising agent for mood disorders that are possibly associated with inflammatory markers [65]. Also, data from TCZ, SAR, and SRK studies demonstrate improvement of pain, fatigue, and mood [66]. However, we do not know whether this is associated with suppression of inflammation in RA or with another direct effect. An ongoing randomized, placebo-controlled, double-blind phase II study (ClinicalTrials.com identifier NCT02473289) is evaluating the efficacy and safety of SRK as adjunctive treatment to monoaminergic antidepressants in adult major depressive disorder patients with a suboptimal response to standard therapy and increased levels of highly sensitive CRP of ≥ 0.3 mg/dL [67].
A human in-vitro model of RA-associated CVD was used to evaluate the impact of various RA treatments (SRK, TCZ, adalimumab [ADA] and tofacitinib) on vessel wall health [68, 69]. Both SRK and TCZ dramatically decreased adhesion molecule and nuclear factor-kB gene expression, while simultaneously increasing vasculo-protective responses, such as endothelial nitric oxide synthase and krüppel-like factor expression, and promoting a contractile smooth muscle cell phenotype. They potently suppressed inflammation and promoted vascular health in this in-vitro model. ADA was a less effective inhibitor of key CVD pathways, while tofacitinib tended to exacerbate CVD pathways. The authors concluded that IL-6 inhibition may provide more CVD benefit compared with drugs targeting other pathways and stated that although broadly comparable, SRK was slightly more potent than TCZ in suppressing vascular inflammation or promoting vascular health. However, this is highly speculative, and it is not possible to differentiate TCZ from SRK based on the available limited safety results.
In a phase I, randomized, double-blind, and placebo-controlled study, SRK was tested in patients with cutaneous or systemic lupus erythematosus [70]. Treatment with intravenous SRK infusions was generally well tolerated and showed linear pharmacokinetics over the dose range studied. SRK suppressed CRP and serum amyloid A concentrations until week 14. However, a multicenter, randomized, double-blind, placebo-controlled phase II study in patients with active lupus nephritis did not confirm efficacy [71]. The median percent change in proteinuria from baseline to week 24 in SRK-treated patients was 0.0%. Of note, the study raised safety concerns since 47.6% of SRK-treated patients experienced one or more serious adverse event (sAE) through week 40. Most of the sAEs were infection-related. Moreover, five patients, all in the SRK treatment group, discontinued the study treatment due to adverse events.
6 Clinical Trials of Agents Targeting the IL-6 Pathway
Strategies of blockade for IL-6 pathway consisted of targeting IL-6R, IL-6, trans-signaling or intracellular signaling.
6.1 IL-6R Blocking
6.1.1 Tocilizumab
This section summarizes the pivotal trials with TCZ in RA. TCZ binds to both the mIL-6R and sIL-6R. In one of the first published studies (SAMURAI), Japanese patients (n = 306) with active RA were randomly assigned to either the TCZ group (8 mg/kg IV, every 4 weeks) or conventional DMARDs group [72]. The TCZ group showed significantly less radiographic disease progression demonstrated by mean total modified Sharp score (TSS). At week 52, the increase in TSS score in the TCZ group (mean difference of 2.3) was significantly lower than that in the DMARDs group (mean difference of 6.1). Besides, the proportions of patients achieving American College of Rheumatology 20%, 50%, and 70% improvement (ACR20, ACR50, and ACR70) were 78, 64, and 44% in the TCZ group and 34, 13, and 6% in DMARDs group, respectively (p < 0.001 for each comparison). Adverse events were more common in the TCZ group compared with the DMARDs group (89% vs 82%; sAEs 18% vs 13%). Most common AEs in the TCZ group were infections and common laboratory abnormalities mostly related to increase in lipid levels.
In another crucial study with TCZ (AMBITION), it was shown for the first time that a biological DMARD (bDMARD) in monotherapy is superior with respect to clinical efficacy over MTX monotherapy in patients with RA for whom previous treatment with MTX/biological agents had not failed [73]. At week 24, TCZ displayed better efficacy than MTX in terms of ACR20 response (69.9% vs 52.5%; p < 0.001), and Disease Activity Score in 28 joints (DAS28) remission rate (33.6% vs 12.1%). The incidence of sAEs with TCZ was 3.8% vs 2.8% with MTX (p = 0.50). Serious infections were reported by four patients in the TCZ and two in the MTX group (1.4% vs 0.7%; p = 0.422). A higher incidence of reversible grade 3 neutropenia (3.1% vs 0.4%) and increased total cholesterol ≥ 240 mg/dL (13.2% vs 0.4%), and a lower incidence of alanine aminotransferase (ALT) elevations > 3 × to < 5 × upper limit of normal (1.0% vs 2.5%) were observed with TCZ compared with MTX.
In a double-blind, randomized, controlled trial (FUNCTION), MTX-naive patients with early progressive RA were randomly assigned (1:1:1:1) to one of four treatment groups: 4 mg/kg TCZ + MTX, 8 mg/kg TCZ + MTX, 8 mg/kg TCZ + placebo, and placebo + MTX [74]. Significantly more patients receiving 8 mg/kg TCZ + MTX and 8 mg/kg TCZ + placebo than receiving placebo + MTX achieved DAS28 remission at week 24 (45% and 39% vs 15%; p < 0.0001). The 8 mg/kg TCZ + MTX group also achieved significantly greater improvement in radiographic disease progression and physical function at week 52 than did patients treated with placebo + MTX. TCZ was effective in combination with MTX and as monotherapy for the treatment of patients with early RA. Two-year results of this study also showed maintenance of these clinical benefits in early RA patients treated with TCZ monotherapy or TCZ + MTX with no new safety signals [75].
To further prove a disease-modifying effect of IL-6R antagonism, the large LITHE study enrolled 1196 patients with moderate to severe RA who had inadequate responses to MTX (MTX-IR). Patients were randomized to receive TCZ (4 or 8 mg/kg, IV) or placebo every 4 weeks in combination with MTX [76]. At week 52, mean change in the Genant-modified Sharp score demonstrated significantly less radiographic progression in the TCZ 8-mg/kg group compared with placebo (0.29 vs 1.13; p < 0.0001). TCZ 8- and 4-mg/kg groups showed improved physical function compared with the placebo group, and proportions of patients achieving ACR20, ACR50, and ACR70 as well as DAS28 remission were higher in those receiving TCZ 8 mg/kg than in those receiving placebo (p < 0.0001 for all comparisons). The safety profile of TCZ was similar to the previous studies showing infections as the most common AEs. Laboratory abnormalities included elevated plasma lipid and hepatic enzyme levels and reduced neutrophil counts. In the extension of this study over 5 years, TCZ + MTX inhibited radiographic progression and maintained improvements in signs and symptoms and physical function in MTX-IR patients with active disease without new safety signals [77].
Finally, the head-to-head study ADACTA was conducted to compare the efficacy and safety of TCZ monotherapy versus ADA monotherapy for the treatment of MTX-IR RA patients who had severe RA for more than 6 months [78]. A total of 326 RA patients were randomly assigned to receive either TCZ (8 mg/kg IV every 4 weeks) or ADA (40 mg SC every 2 weeks). Of note, mean DAS28 improvement was significantly higher in the TCZ (− 3.3) than in the ADA group (− 1.8) (difference − 1.5, 95% CI − 1.8 to −1.1; p < 0.0001) from baseline to week 24, while safety findings were comparable between the treatment arms. Sixteen of 162 (10%) patients in the ADA group versus 19 of 162 (12%) in the TCZ group had sAEs. More patients in the TCZ group than in the ADA group showed an increase in LDL-cholesterol, ALT levels, as well as decreased platelet and neutrophil counts.
6.1.2 Sarilumab
SAR, like TCZ, also binds to both the mIL-6R and sIL-6R. However, SAR differs from TCZ in structure and affinity [79]. SAR is the first fully human mAb against IL-6Rα and showed 10- to 40-fold greater affinity to recombinant monomeric human and monkey IL-6R compared with TCZ in a preclinical study [80]. The conducted phase II and phase III trials demonstrated efficacy of SAR both in MTX-IR and tumor necrosis factor inadequate responder (TNF-IR)-active RA patients [81,82,83,84]. In addition, SAR monotherapy demonstrated a clear superiority over ADA monotherapy in patients with intolerance or inadequate response to MTX [85]. Moreover, although displaying a significantly higher affinity and longer half-life, SAR showed a similar safety profile compared with TCZ [86]. Taken together, these results led to approval of the compound for treatment of RA by the European Medicines Agency (EMA) and US Food and Drug Administration (FDA) in 2017.
6.1.3 ALX-0061
ALX-0061 (vobarilizumab) is a bispecific anti-IL-6R nanobody engineered to have an extended half-life in vivo by targeting human serum albumin, in combination with strong target binding using a single anti-IL-6R building block. It seems to modulate the IL-6 trans-signaling pathway rather than the classical mIL-6R-dependent pathway. In fact, these kinds of molecules are designed according to a new type of immunoglobulin, which was initially discovered in the serum of camels (Camelus dromedarius), consisting of only heavy-chain dimers and called ‘heavy-chain-only antibodies’ (HCAbs) [87]. They show excellent tissue distribution and high temperature and pH stability. Recombinant HCAbs are easy to produce and can be converted into different formats such as Fc-fusion proteins or hetero-dimers [88]. Due to their small size, these nanobodies have low toxicity and immunogenicity in vivo, they show relatively high sequence identity to human heavy chain variable domain VH, and are rapidly cleared via the kidney [87, 89, 90]. Affinity of ALX-0061 to sIL-6R is 2400-fold and to mIL-6R is 17-fold higher than TCZ. The data from a phase I/II study were promising with ACR20 response rates up to 84% and DAS28 remission rates up to 58% [91]. In a phase IIb monotherapy study (head-to-head vs TCZ) in 251 RA patients, vobarilizumab demonstrated a high rate of clinical remission based on DAS28-CRP in up to 41% of patients, as compared with 27% of patients with TCZ (ClinicalTrials.com identifier NCT02287922) [92].
6.2 IL-6 Blocking
6.2.1 Siltuximab
Siltuximab is an anti-IL-6 chimeric IgG1κ mAb that binds to IL-6 with high affinity, thus neutralizing the cytokine bioactivity and inhibiting B-cell proliferation [93,94,95]. Clinical trials of siltuximab have demonstrated significant efficacy and tolerance in patients with idiopathic MCD, leading to FDA approval. In a systematic review of 171 cases of MCD patients treated with siltuximab, while traditional treatment methods were able to achieve a 5-year survival rate of only 55–77%, results of siltuximab treatment demonstrated 5-year survival rates of nearly 96.4% (only two deaths reported out of 55 patients with follow-up data) [96].
6.2.2 MEDI5117
A fully human mAb targeting IL-6, MEDI5117 has been developed by variable domain engineering to achieve higher affinity and improved half-life from the progenitor anti-IL-6 human mAb CAT6001 [97]. However, a phase I trial assessing the safety and tolerability of MEDI5117 in RA patients has been terminated due to difficulties with patient recruitment (ClinicalTrials.com identifier NCT01559103) [98].
6.2.3 Clazakizumab
Clazakizumab (BMS945429; ALD518), another humanized mAb, binds to circulating IL-6 cytokine rather than the IL-6 receptor, blocking both classic signaling and trans-signaling [99]. In a study comparing the potential of clazakizumab and tocilizumab with multiple in-vitro assays for IL-6-induced functions, clazakizumab was between 3- and 120-fold more potent than TCZ in vitro for inhibiting signaling, proliferation, activation, antibody production, and secretion of acute phase protein [100]. In RA patients with an inadequate response to MTX, clazakizumab treatment either as monotherapy or in combination with MTX was well tolerated and associated with significant improvements in disease activity compared with MTX therapy alone [101]. AEs and laboratory abnormality profiles were consistent with the class effect of IL-6 blockade. However, no clear dose-dependent clinical effect was observed and phase III trials have not been performed following phase II.
6.2.4 Olokizumab
Olokizumab (OKZ, CDP6038) is a humanized IgG4 mAb specific for IL-6. It has been shown to be well tolerated in healthy volunteers [102]. The bioavailability of the SC doses ranged from 84.2 to 92.5% with a mean terminal half-life of 31.5 days. Rapid decreases in CRP concentrations were observed, without any dose dependency. In a dose-ranging, double-blind study in RA patients with moderate-to-severe disease activity who had previously failed anti-tumor necrosis factor (anti-TNF) therapy, OKZ produced significantly greater reductions in DAS28-CRP from baseline at Week 12 compared with placebo. OKZ treatment, at several doses, demonstrated similar efficacy and a comparable safety profile to TCZ. Reported AEs were consistent with class effect of IL-6 blockade [103]. In RA patients receiving MTX who had previously failed anti-TNF therapy, OKZ treatment of both Western and Asian patients with moderate to severe RA resulted in sustained and similar levels of improvement across a range of patient-related outcomes in both populations in the randomized controlled trial and open label [103,104,105]. The drug is currently in phase III with various key objectives and a long-term extension study [106,107,108,109] (ClinicalTrials.com identifiers NCT02760433, NCT02760368, NCT02760407, NCT03120949). The reported death rates from the open-label extension of two studies, which enrolled Western and Asian RA patients who had failed previous anti-TNF therapy, were 1.1% and 0%, respectively [110,111,112].
6.2.5 Sirukumab
SRK is a human anti-IL-6 mAb that binds to IL-6 with high affinity and specificity and prevents IL-6 from binding to both membranous and soluble forms of IL-6R. Of note, SRK is an IgG1κ mAb in contrast to olokizumab, an IgG4 antibody. Since IgG4 antibodies have low affinity for Fc-γ-receptors and C1q compared with other IgG subclasses, they have minimal ability to activate cells or initiate complement activation [113]. The structural differences of these two drugs theoretically may lead to some variations in their efficacy and safety.
In a randomized phase II study, the safety and efficacy of SRK was evaluated in MTX-IR patients with active RA [48]. The study consisted of two parts; in part A (proof of concept), 36 patients with RA were randomized to SC placebo or SRK 100 mg every 2 weeks (q2w) through week 10, with crossover treatment during weeks 12–22. In part B (dose finding), 151 patients with RA were randomized to five arms, SC SRK (100 mg q2w, 100 mg q4w, 50 mg q4w, or 25 mg q4w) for 24 weeks, or placebo through week 10, with crossover to SRK 100 mg every 2 weeks during weeks 12–24. SRK 100 mg every 2 weeks was associated with greater improvements in mean DAS28-CRP at week 12 and the other primary efficacy outcome—ACR50 response at week 12—was only achieved with SRK 100 mg every 2 weeks. Changes in neutrophil and platelet counts, transient elevations in ALT and an increase in lipid levels were observed consistent with other IL-6 pathway targeting strategies. Through week 12, the incidence of AEs for SRK was similar with placebo-treated patients. Infections were the most common type of AE, however no opportunistic infections were reported. One death occurred in Part B (SRK 100 mg q2w) unrelated to study drug (brain aneurysm rupture).
Subsequently, SRK has also been investigated in five phase III trials. These included patients with active RA and at least one poor prognostic factor refractory to DMARDs (including MTX or sulfasalazine) (ARA3001, ARA3002), and patients with moderately to severely active RA who were unresponsive or intolerant to anti-TNF agents (ARA3003) [114,115,116]. SRK showed significant improvements in RA symptoms along with inhibition of structural damage progression and improvements in quality of life in these studies. In addition, a head-to-head study with ADA has been completed in biologically naive, moderately to severely active RA patients for whom their physicians have judged that MTX therapy would not be appropriate (ARA3005) [117]. Patients treated with SRK monotherapy showed greater improvements in DAS28 and similar ACR50 response rates compared with ADA monotherapy. For patients who completed the ARA3002 and ARA3003, an extension study to evaluate the long-term safety and efficacy of SRK in RA was performed (ARA3004) [118]. Table 1 shows the phase II and phase III studies of SRK in patients with RA.
7 Sirukumab—Mortality Rates
The clinical studies performed to date showed largely similar clinical efficacy data for SRK compared with TCZ and other IL-6 pathway targeting agents. Furthermore, the drug safety profile seemed to be consistent with a class effect showing similar adverse effects and laboratory abnormality profiles. However, death rates in SRK arms compared with placebo, especially in the controlled period, raised safety concerns, which led to the decision by the FDA to decline the approval of SRK in August 2017.
The majority of the committee (11 to 2) did not agree that the safety profile of SRK 50 mg SC every 4 weeks is adequate to support the approval of SRK for the treatment of adult patients with moderately to severely active RA who have had an inadequate response or are intolerant to one or more DMARDs [119]. The majority of the committee agreed that it is unclear whether the imbalance in all-cause mortality is a true safety signal or whether it is a result of bias due to the study design. The majority of the committee also agreed that additional studies should be conducted to further define the safety profile of SRK. One committee member proposed that the sponsor should find alternative methods to reanalyzing the data in addition to conducting more studies. One of the members who voted ‘yes’ stated that SRK is no less safe than the other approved biologics on the market. The majority agreed that efficacy was clear but the safety data was lacking, and a few members emphasized that SRK would be more suitable for patients who have had an inadequate response or are intolerant to one or more biologic DMARDs. These members added that the benefit of this drug for this narrower indication might outweigh the unknown safety risks, especially for patients who have limited treatment options left.
Most common causes of mortality were major cardiovascular events (MACE), infections, and malignancies. In studies ARA3002 and ARA3003, one death occurred in each treatment group through the 18-week placebo-controlled period. Afterwards, an additional eight deaths occurred in patients randomized to SRK 50 mg every 4 weeks (n = 3) and in patients randomized to SRK 100 mg every 2 weeks (n = 5) through 52 weeks of exposure. Up until the summary of clinical safety (SCS) cutoff date (02 February 2016), a total of 29 deaths were reported from studies ARA3002 and ARA3003, including additional exposure periods during the ARA3004 long-term extension [120].
A single fatality was reported in the group that received SRK 100 mg every 2 weeks in the ARA3005 study and an additional eight deaths were reported until 29 July 2016 as the cutoff date for the 120-day safety update. Thus, the mortality rates in the phase III study programs (ARA3001, ARA3002, ARA3003, ARA3004, and ARA3005) to the 120-day safety update cutoff date were 0.68 and 0.71 deaths per 100 patient-years, respectively, in patients receiving SRK 50 mg every 4 weeks and 100 mg every 2 weeks [121]. Additional deaths (one death under the SRK 50 mg q4w group occurred during the safety update period but lately reported after the 29 July 2016 cutoff, four deaths in the phase III studies beyond the 16-week follow-up after the last study dose and one death in the phase II study [C1377T04]) have not been included in the above incidence rates (Fig. 2).

Rate of deaths per 100 patient-years. Pbo-Controlled refers to 18-week placebo-controlled periods of studies ARA3002 and ARA3003, Week 52 refers to 18-week placebo-controlled period as well as data beyond Week 18 and through Week 52 of studies ARA3002 and ARA3003 only, SCS Cutoff refers to studies ARA3002 and ARA3003 and their long-term extension study ARA3004 through the SCS Cutoff date of 02 February 2016, and 120-day Cutoff refers to all studies (ARA3001, ARA3002, ARA3003, ARA3004, and ARA3005) through a cutoff date of 29 July 2016. Beyond the 16-week safety follow-up window after the last dose of study agent for all the phase III studies, four more deaths occurred: two in ARA3002, one in ARA3003, and one in ARA3005. These patients are not included in the figure. In addition, one death occurred in the phase II study C1377T04. This patient is also not included in the figure. [Reproduced from the FDA webpage showing an Arthritis Advisory Committee Briefing Document by Janssen Research & Development for Plivensia™ (sirukumab). The abbreviation for sirukumab has been changed from the original figure] [120]. PBO placebo, PY(Pt-Yrs) patient years, SRK sirukumab
Mortality rates determined by 6-month increments of exposure to SRK showed no increase or dose effect in mortality rates with prolonged exposure (Fig. 3) [122]. Moreover, the rates of events for the most common causes of mortality (MACE, infections, and malignancies) remained constant over time.

Analysis of mortality incidence rates over time. Incidence rate (based on 100 subject-years of follow-up) of deaths in 6-month incremental periods with sirukumab treatment exposure time aligned to Week 0 for early escape (EE), late escape (LE), and crossover (CO) subjects; all subjects in phase III studies. [Reproduced from the EMA webpage showing the withdrawal assessment report for Plivensia™ (sirukumab)] [122]
Of note, mortality rates in SRK-treated patients were consistent with mortality rates previously reported in the general RA population [123,124,125]. Also when compared with other randomized clinical trials in RA populations, mortality rates were similar. Regarding placebo-treated patients, due to smaller cumulative follow-up periods with placebo-treated patients, more variability was reported across RA development programs (Fig. 4) [122].

Exposure-adjusted mortality rates from the SRK RCT program and published rates from comparator RCT programs. Death rates in the sirukumab program were compared indirectly against publicly reported, exposure-adjusted mortality rates from development programs or large, randomized, controlled clinical trials of approved RA therapies. The combined sirukumab mortality rate per 100 subject-years of exposure of 0.66 (95% CI 0.44–0.95) in the long-term phase III studies (i.e., ARA3001, ARA3002, ARA3003, and ARA3005, as well as ARA3002 and ARA3003 subjects in ARA3004) and the placebo mortality rate of 0.19 (95% CI 0.00–1.07) for the placebo-controlled studies (ARA3002, ARA3003) were plotted beside the exposure-adjusted mortality rates from the comparator programs and trials. The mortality rate for both placebo- and sirukumab-treated patients falls within the range for the mortality rates of other RCT populations. The point estimate of the placebo rate is on the lower end of the range of mortality rates reported for placebo groups in various clinical trials, albeit with wide 95% confidence intervals. [Reproduced from the EMA webpage showing withdrawal assessment report for Plivensia™ (sirukumab)] [122]. IV intravenous, Pbo placebo, PY(Pt-Yrs) patient years, RA rheumatoid arthritis, RCT randomized controlled trials, SC subcutaneous, SRK sirukumab
The overall safety analysis showed an imbalance in terms of death rates between placebo- and SRK-treated patients; however, this observation is confounded, since placebo subjects with a response of < 20% in swollen and tender joints also received the active drug from week 18 (ARA3002 and ARA3003) or 40 (ARA3002) “early escape (EE) or late escape (LE)”. In the 18-week, true placebo-controlled period, mortality rates were identical in the placebo- and SRK-treated patients. Comparisons after week 18 may be confounded by some factors, such as different health status for patients remaining in the placebo group compared with switchers. Also, the ‘crossover’ design (all placebo-treated subjects in ARA3002 were re-randomized at week 52 and all placebo-treated subjects in ARA3003 were re-randomized at week 24) resulted in various treatment groups with varying drug-exposure periods. From the statistical point of view, given the few number of events, an exposure-adjusted comparison is also not fully reliable. On the other hand, although mortality rates in SRK trials are consistent with previous RA studies and RA cohorts, such inter-study comparisons must be interpreted with great caution. The limited placebo exposure relative to SRK exposure makes interpretation of mortality rates difficult. Thus, due to these methodical limitations, no clear conclusion can be made with respect to the safety profile and especially risk of death under SRK to date.
8 Concluding Remarks
In the last decade, the importance of this cytokine has risen with the successful introduction of IL-6 inhibitors TCZ and SAR for the treatment of rheumatoid arthritis. The high potency and favorable efficacy/safety profile of these agents encouraged investigation into targeting different points of this pathway. In this context, on the basis of less circulating cytokine versus receptor, polymorphisms in the IL-6 receptor gene and the fact that the IL-6 receptor has additional ligands such as CNTF and p28 led to the hypothesis that IL-6 inhibition may have additional advantages over IL-6R inhibition, such as lower drug load, longer half-life, and more specific and efficacious responses.
However, no anti-IL6 antibody has been approved for RA so far and some efficacy and safety results are inconsistent or inconclusive. The currently available results imply that the IL-6 pathway may be more complex than supposed and targeting different points of this pathway may bring some risks beyond estimated advantages. Currently, we do not know whether the imbalance in mortality rates seen for SRK is a true safety signal or a result of bias due to the study design. Therefore, further long-term clinical data as well as basic research is needed to allow a deeper insight into IL-6 signaling as a crucial cytokine both in inflammation and regulation of autoimmunity.
References
Schett G. Physiological effects of modulating the interleukin-6 axis. Rheumatology (Oxford). 2018;57:ii43-ii50.
Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16:448–57.
Calabrese LH, Rose-John S. IL-6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol. 2014;10:720–7.
Yao X, Huang J, Zhong H, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141:125–39.
Hirano T. Revisiting the 1986 molecular cloning of interleukin 6. Front Immunol. 2014;5:456.
Naka T, Nishimoto N, Kishimoto T. The paradigm of IL-6: from basic science to medicine. Arthritis Res. 2002;4(Suppl 3):S233–42.
Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF. How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat Med. 2013;19:822–4.
Bethin KE, Vogt SK, Muglia LJ. Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc Natl Acad Sci USA. 2000;97:9317–22.
Kraakman MJ, Kammoun HL, Allen TL, et al. Blocking IL-6 trans-signaling prevents high-fat diet-induced adipose tissue macrophage recruitment but does not improve insulin resistance. Cell Metab. 2015;21:403–16.
Rose-John S, Winthrop K, Calabrese L. The role of IL-6 in host defence against infections: immunobiology and clinical implications. Nat Rev Rheumatol. 2017;13:399–409.
Schaper F, Rose-John S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015;26:475–87.
Lehmann U, Schmitz J, Weissenbach M, et al. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem. 2003;278:661–71.
Taga T, Hibi M, Hirata Y, et al. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989;58:573–81.
Mullberg J, Schooltink H, Stoyan T, et al. The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol. 1993;23:473–80.
Heink S, Yogev N, Garbers C, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol. 2017;18:74–85.
Scheller J, Rose-John S. The interleukin 6 pathway and atherosclerosis. Lancet. 2012;380:338.
Scheller J, Garbers C, Rose-John S. Interleukin-6: from basic biology to selective blockade of pro-inflammatory activities. Semin Immunol. 2014;26:2–12.
Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest. 2011;121:3375–83.
Rafiq S, Frayling TM, Murray A, et al. A common variant of the interleukin 6 receptor (IL-6r) gene increases IL-6r and IL-6 levels, without other inflammatory effects. Genes Immun. 2007;8:552–9.
Ferreira RC, Freitag DF, Cutler AJ, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013;9:e1003444.
Jostock T, Mullberg J, Ozbek S, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem. 2001;268:160–7.
Atreya R, Mudter J, Finotto S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–8.
Hurst SM, Wilkinson TS, McLoughlin RM, et al. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity. 2001;14:705–14.
Nowell MA, Richards PJ, Horiuchi S, et al. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J Immunol. 2003;171:3202–9.
Mitsuyama K, Matsumoto S, Rose-John S, et al. STAT3 activation via interleukin 6 trans-signalling contributes to ileitis in SAMP1/Yit mice. Gut. 2006;55:1263–9.
Nowell MA, Williams AS, Carty SA, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613–22.
Matsumoto S, Hara T, Mitsuyama K, et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J Immunol. 2010;184:1543–51.
Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501.
Doganci A, Eigenbrod T, Krug N, et al. The IL-6R alpha chain controls lung CD4 + CD25 + Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–25.
Hoge J, Yan I, Janner N, et al. IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J Immunol. 2013;190:703–11.
Sodenkamp J, Waetzig GH, Scheller J, et al. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology. 2012;217:996–1004.
Safety and efficacy of TJ301 IV in participants with active ulcerative colitis. from https://clinicaltrials.gov/ct2/show/NCT03235752?cond=FE+999301&rank=1. Retrieved 27 May 2018.
Dasgupta B, Corkill M, Kirkham B, Gibson T, Panayi G. Serial estimation of interleukin 6 as a measure of systemic disease in rheumatoid arthritis. J Rheumatol. 1992;19:22–5.
Robak T, Gladalska A, Stepien H, Robak E. Serum levels of interleukin-6 type cytokines and soluble interleukin-6 receptor in patients with rheumatoid arthritis. Mediators Inflamm. 1998;7:347–53.
Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988;31:784–8.
Madhok R, Crilly A, Watson J, Capell HA. Serum interleukin 6 levels in rheumatoid arthritis: correlations with clinical and laboratory indices of disease activity. Ann Rheum Dis. 1993;52:232–4.
Straub RH, Muller-Ladner U, Lichtinger T, et al. Decrease of interleukin 6 during the first 12 months is a prognostic marker for clinical outcome during 36 months treatment with disease-modifying anti-rheumatic drugs. Br J Rheumatol. 1997;36:1298–303.
Atsumi T, Ishihara K, Kamimura D, et al. A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J Exp Med. 2002;196:979–90.
Sasai M, Saeki Y, Ohshima S, et al. Delayed onset and reduced severity of collagen-induced arthritis in interleukin-6-deficient mice. Arthritis Rheum. 1999;42:1635–43.
Hata H, Sakaguchi N, Yoshitomi H, et al. Distinct contribution of IL-6, TNF-alpha, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest. 2004;114:582–8.
Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. 2018;17:395–412.
Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–42.
Briso EM, Dienz O, Rincon M. Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J Immunol. 2008;180:7102–6.
Dominitzki S, Fantini MC, Neufert C, et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4 + CD25 T cells. J Immunol. 2007;179:2041–5.
Thiolat A, Semerano L, Pers YM, et al. Interleukin-6 receptor blockade enhances CD39 + regulatory T cell development in rheumatoid arthritis and in experimental arthritis. Arthritis Rheumatol. 2014;66:273–83.
Sirukumab Presentation to the Arthritis Advisory Committee August 2, 2017 Janssen R&D, LLC. https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/arthritisadvisorycommittee/ucm570357.pdf. Retrieved 27 May 2018.
Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B. Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2014;73:1616–25.
Burmester GR, Rubbert-Roth A, Cantagrel A, et al. A randomised, double-blind, parallel-group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease-modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study). Ann Rheum Dis. 2014;73:69–74.
Kivitz A, Olech E, Borofsky M, et al. Subcutaneous tocilizumab versus placebo in combination with disease-modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66:1653–61.
Abdallah H, Hsu JC, Lu P, et al. Pharmacokinetic and pharmacodynamic analysis of subcutaneous tocilizumab in patients with rheumatoid arthritis from 2 randomized, controlled trials: SUMMACTA and BREVACTA. J Clin Pharmacol. 2017;57:459–68.
Enevold C, Baslund B, Linde L, et al. Interleukin-6-receptor polymorphisms rs12083537, rs2228145, and rs4329505 as predictors of response to tocilizumab in rheumatoid arthritis. Pharmacogenet Genomics. 2014;24:401–5.
Schuster B, Kovaleva M, Sun Y, et al. Signaling of human ciliary neurotrophic factor (CNTF) revisited. The interleukin-6 receptor can serve as an alpha-receptor for CTNF. J Biol Chem. 2003;278:9528–35.
Garbers C, Spudy B, Aparicio-Siegmund S, et al. An interleukin-6 receptor-dependent molecular switch mediates signal transduction of the IL-27 cytokine subunit p28 (IL-30) via a gp130 protein receptor homodimer. J Biol Chem. 2013;288:4346–54.
Miller RG, Bryan WW, Dietz MA, et al. Toxicity and tolerability of recombinant human ciliary neurotrophic factor in patients with amyotrophic lateral sclerosis. Neurology. 1996;47:1329–31.
Petes C, Mintsopoulos V, Finnen RL, Banfield BW, Gee K: The effects of CD14 and IL-27 on induction of endotoxin tolerance in human monocytes and macrophages. J Biol Chem. 2018.
Kalliolias GD, Gordon RA, Ivashkiv LB. Suppression of TNF-alpha and IL-1 signaling identifies a mechanism of homeostatic regulation of macrophages by IL-27. J Immunol. 2010;185:7047–56.
Petes C, Mariani MK, Yang Y, Grandvaux N, Gee K. Interleukin (IL)-6 inhibits IL-27- and IL-30-mediated inflammatory responses in human monocytes. Front Immunol. 2018;9:256.
Zhang C, Xin H, Zhang W, et al. CD5 binds to interleukin-6 and induces a feed-forward loop with the transcription factor STAT3 in B cells to promote cancer. Immunity. 2016;44:913–23.
McFarland-Mancini MM, Funk HM, Paluch AM, et al. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J Immunol. 2010;184:7219–28.
Lazzerini PE, Capecchi PL, Guidelli GM, et al. Spotlight on sirukumab for the treatment of rheumatoid arthritis: the evidence to date. Drug Des Devel Ther. 2016;10:3083–98.
Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Matcham F, Rayner L, Steer S, Hotopf M. The prevalence of depression in rheumatoid arthritis: a systematic review and meta-analysis. Rheumatology (Oxford). 2013;52:2136–48.
Hsu B, Wang D, Sun Y, Chen G. Improvement in measures of depressed mood and anhedonia, and fatigue, in a randomized, placebo-controlled, phase 2 study of sirukumab, a human anti-interleukin-6 antibody, in patients with rheumatoid arthritis. Ann Rheum Dis. 2015;74(Suppl 2):720–1. https://doi.org/10.1136/annrheumdis-2015-eular.4081.
Zhou AJ, Lee Y, Salvadore G, et al. Sirukumab: a potential treatment for mood disorders? Adv Ther. 2017;34:78–90.
Choy EHS, Calabrese LH: Neuroendocrine and neurophysiological effects of interleukin 6 in rheumatoid arthritis. Rheumatology (Oxford). 2017.
An efficacy and safety study of sirukumab in participants with major depressive disorder. https://clinicaltrials.gov/ct2/results?cond=&term=NCT02473289&cntry=&state=&city=&dist=. Retrieved 27 May 2018.
Feaver R, Collado S, Hoang S, et al. The anti-IL-6 antibody sirukumab inhibits vascular inflammation in a human surrogate model of atherosclerosis. Abstract number: 439, ACR/ARHP annual meeting, 2014.
Feaver R, Collado S, Hoang S, et al. Neutralization of IL6 by sirukumab (SIR) inhibits inflammation and cellular stress in a human vascular surrogate system of atherosclerosis. Ann Rheum Dis. 2015;74:444–5. https://doi.org/10.1136/annrheumdis-2015-eular.5132.
Szepietowski JC, Nilganuwong S, Wozniacka A, et al. Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum. 2013;65:2661–71.
Rovin BH, van Vollenhoven RF, Aranow C, et al. A multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of treatment with sirukumab (CNTO 136) in patients with active lupus nephritis. Arthritis Rheumatol. 2016;68:2174–83.
Nishimoto N, Hashimoto J, Miyasaka N, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an X ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis. 2007;66:1162–7.
Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69:88–96.
Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis. 2016;75:1081–91.
Burmester GR, Rigby WF, van Vollenhoven RF, et al. Tocilizumab combination therapy or monotherapy or methotrexate monotherapy in methotrexate-naive patients with early rheumatoid arthritis: 2-year clinical and radiographic results from the randomised, placebo-controlled FUNCTION trial. Ann Rheum Dis. 2017;76:1279–84.
Kremer JM, Blanco R, Brzosko M, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011;63:609–21.
Kremer JM, Blanco R, Halland AM, et al. Clinical efficacy and safety maintained up to 5 years in patients with rheumatoid arthritis treated with tocilizumab in a randomised trial. Clin Exp Rheumatol. 2016;34:625–33.
Gabay C, Emery P, van Vollenhoven R, et al. Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA): a randomised, double-blind, controlled phase 4 trial. Lancet. 2013;381:1541–50.
Kim GW, Lee NR, Pi RH, et al. IL-6 inhibitors for treatment of rheumatoid arthritis: past, present, and future. Arch Pharm Res. 2015;38:575–84.
Rafique A, Martin J, Blome M, Huang T, Ouyang A, Papadopoulos N. Evaluation of the binding kinetics and functional bioassay activity of sarilumab and tocilizumab to the human IL-6 receptor (IL-6R) alpha. Ann Rheum Dis. 2013;72(Suppl3):797.
Huizinga TW, Fleischmann RM, Jasson M, et al. Sarilumab, a fully human monoclonal antibody against IL-6Ralpha in patients with rheumatoid arthritis and an inadequate response to methotrexate: efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann Rheum Dis. 2014;73:1626–34.
Genovese MC, Fleischmann R, Kivitz AJ, et al. Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III study. Arthritis Rheumatol. 2015;67:1424–37.
Strand V, Kosinski M, Chen CI, et al. Sarilumab plus methotrexate improves patient-reported outcomes in patients with active rheumatoid arthritis and inadequate responses to methotrexate: results of a phase III trial. Arthritis Res Ther. 2016;18:198.
Fleischmann R, van Adelsberg J, Lin Y, et al. Sarilumab and nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis and inadequate response or intolerance to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2017;69:277–90.
Burmester GR, Lin Y, Patel R, et al. Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): a randomised, double-blind, parallel-group phase III trial. Ann Rheum Dis. 2017;76:840–7.
Raimondo MG, Biggioggero M, Crotti C, Becciolini A, Favalli EG. Profile of sarilumab and its potential in the treatment of rheumatoid arthritis. Drug Des Devel Ther. 2017;11:1593–603.
Hamers-Casterman C, Atarhouch T, Muyldermans S, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–8.
Rissiek B, Koch-Nolte F, Magnus T. Nanobodies as modulators of inflammation: potential applications for acute brain injury. Front Cell Neurosci. 2014;8:344.
Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–97.
Van Roy M, Ververken C, Beirnaert E, et al. The preclinical pharmacology of the high affinity anti-IL-6R nanobody(R) ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res Ther. 2015;17:135.
Holz JB, Sargentini-Maier L, De Bruyn S, Gachályi B, Udvaros I, Rojkovich B, Bruk S, Sramek P, Korkosz M, Krause K, Schoen P, D’Artois J, Verschueren K, Willems W, De Swert K, Arold G. Twenty-four weeks of treatment with a novel anti-IL-6 receptor nanobody® (aALX-0061) resulted in 84% ACR20 improvement and 58% DAS28 remission in a phase I/II study in RA. Ann Rheum Dis. 2013;72:A64.
http://www.ablynx.com/rd-portfolio/clinical-programmes/vobarilizumab/. Retrieved 07 June 2018.
Mayer CL, Xie L, Bandekar R, et al. Dose selection of siltuximab for multicentric Castleman’s disease. Cancer Chemother Pharmacol. 2015;75:1037–45.
van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15:966–74.
van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol. 2010;28:3701–8.
Sitenga J, Aird G, Ahmed A, Silberstein PT. Impact of siltuximab on patient-related outcomes in multicentric Castleman’s disease. Patient Relat Outcome Meas. 2018;9:35–41.
Finch DK, Sleeman MA, Moisan J, et al. Whole-molecule antibody engineering: generation of a high-affinity anti-IL-6 antibody with extended pharmacokinetics. J Mol Biol. 2011;411:791–807.
Study to assess the safety and tolerability of MEDI5117 in rheumatoid arthritis patients. https://clinicaltrials.gov/ct2/results?cond = &term = NCT01559103&cntry = &state = &city = &dist=. Retrieved 28 May 2018.
Mease P, Strand V, Shalamberidze L, et al. A phase II, double-blind, randomised, placebo-controlled study of BMS945429 (ALD518) in patients with rheumatoid arthritis with an inadequate response to methotrexate. Ann Rheum Dis. 2012;71:1183–9.
Zhao Q, Pang J, Shuster D, Hung C, Baglino S, Dodge R, et al. Anti-IL-6 antibody clazakizumab is more potent than tocilizumab in blocking in vitro and ex vivo IL-6-induced functions (abstract). Arthritis Rheum. 2013;65(Suppl):S1020.
Weinblatt ME, Mease P, Mysler E, et al. The efficacy and safety of subcutaneous clazakizumab in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate: results from a multinational, phase IIb, randomized, double-blind, placebo/active-controlled, dose-ranging study. Arthritis Rheumatol. 2015;67:2591–600.
Kretsos K, Golor G, Jullion A, et al. Safety and pharmacokinetics of olokizumab, an anti-IL-6 monoclonal antibody, administered to healthy male volunteers: a randomized phase I study. Clin Pharmacol Drug Dev. 2014;3:388–95.
Genovese MC, Fleischmann R, Furst D, et al. Efficacy and safety of olokizumab in patients with rheumatoid arthritis with an inadequate response to TNF inhibitor therapy: outcomes of a randomised Phase IIb study. Ann Rheum Dis. 2014;73:1607–15.
Genovese MC, Durez P, Fleischmann R, Tanaka Y, Furst DE, Yamanaka H, Vasyutin I, Kaviarasu T, Korneva E, Koloda D, Takeuchi T. Olokizumab treatment of both Western and Asian patients with rheumatoid arthritis who have failed anti-TNF treatment results in sustained improvements in patient-reported outcomes (abstract). Arthritis Rheumatol. 2016;68 :10.
Takeuchi T, Tanaka Y, Yamanaka H, et al. Efficacy and safety of olokizumab in Asian patients with moderate-to-severe rheumatoid arthritis, previously exposed to anti-TNF therapy: results from a randomized phase II trial. Mod Rheumatol. 2016;26:15–23.
Evaluation of the effectiveness and safety of two dosing regimens of olokizumab (OKZ), compared to placebo, in subjects with rheumatoid arthritis (RA) who are taking an existing medication called a tumour necrosis factor alpha inhibitor but have active disease. https://clinicaltrials.gov/ct2/results?cond=&term = NCT02760433&cntry = &state = &city = &dist=. Retrieved 28 May 2018.
Evaluation of the effectiveness and safety of two dosing regimens of olokizumab (OKZ), compared to placebo, in subjects with rheumatoid arthritis (RA) who are taking methotrexate but have active disease. from https://clinicaltrials.gov/ct2/results?cond=&term=NCT02760368&cntry=&state=&city=&dist=. Retrieved 28 May 2018.
Evaluation of the effectiveness and safety of two dosing regimens of olokizumab (OKZ), compared to placebo and adalimumab, in subjects with rheumatoid arthritis (RA) who are taking methotrexate but have active disease. https://clinicaltrials.gov/ct2/results?cond=&term=NCT02760407&cntry=&state=&city=&dist=. Retrieved 28 May 2018.
Evaluation of the long term safety, tolerability and efficacy of two dosing regimens of olokizumab (OKZ), in subjects with rheumatoid arthritis (RA) who previously completed 24 weeks of blinded treatment in one of the core studies—CREDO 1, 2 or 3. https://clinicaltrials.gov/ct2/results?cond=&term=NCT03120949&cntry=&state=&city=&dist=. Retrieved 28 May 2018.
Genovese MC, Fleischmann R, Tanaka Y, Furst DE, Yamanaka H, Joshi R, Zhu W, Shao J, Mashimo H, Takeuchi T. Long-term safety and efficacy of olokizumab in patients with moderate-to-severe rheumatoid arthritis who have previously failed anti-TNF treatment. ACR/ARHP annual meeting, 2015.
Open-label study to assess the safety and efficacy of CDP6038 in patients who completed RA0056. https://clinicaltrials.gov/ct2/show/NCT01296711?term=NCT01296711&rank=1. Retrieved 23 Oct 2018.
The long-term safety and efficacy of olokizumab (CDP6038) with active rheumatoid arthritis. from https://clinicaltrials.gov/ct2/show/study/NCT01533714?term=NCT01533714&rank=1. Retrieved 23 Oct 2018.
Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520.
Takeuchi T, Yamanaka H, Harigai M, et al. Sirukumab in rheumatoid arthritis refractory to sulfasalazine or methotrexate: a randomized phase 3 safety and efficacy study in Japanese patients. Arthritis Res Ther. 2018;20:42.
Takeuchi T, Thorne C, Karpouzas G, et al. Sirukumab for rheumatoid arthritis: the phase III SIRROUND-D study. Ann Rheum Dis. 2017;76:2001–8.
Aletaha D, Bingham CO 3rd, Tanaka Y, et al. Efficacy and safety of sirukumab in patients with active rheumatoid arthritis refractory to anti-TNF therapy (SIRROUND-T): a randomised, double-blind, placebo-controlled, parallel-group, multinational, phase 3 study. Lancet. 2017;389:1206–17.
Taylor PC, Schiff MH, Wang Q, et al. Efficacy and safety of monotherapy with sirukumab compared with adalimumab monotherapy in biologic-naive patients with active rheumatoid arthritis (SIRROUND-H): a randomised, double-blind, parallel-group, multinational, 52-week, phase 3 study. Ann Rheum Dis. 2018;77:658–66.
Long-term safety and efficacy of sirukumab in participants with RA completing studies CNTO136ARA3002 or CNTO136ARA3003. https://clinicaltrials.gov/ct2/results?cond=&term=NCT01856309&cntry=&state=&city=&dist=. Retrieved 29 May 2018.
Food and Drug Administration, Center for drug evaluation and research, summary minutes of the arthritis advisory committee meeting, 2 August 2017. https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/arthritisadvisorycommittee/ucm575678.pdf. Retrieved 29 May 2018.
Arthritis Advisory Committee briefing document by Janssen Research & Development for Plivensia™ (sirukumab), 28 June 2017. https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/arthritisadvisorycommittee/ucm569153.pdf. Retrieved 29 May 2018.
Aletaha D, Thorne C, Schiff M, Harigai M, Agarwal P, Rao R, Cohen C, Cheng B, Brown K, Hsu B. Integrated phase 3 safety results of sirukumab, an anti-IL-6 cytokine monoclonal antibody, in patients with active rheumatoid arthritis (abstract). Arthritis Rheumatol. 2017; 69 (suppl 10). http://acrabstracts.org/abstract/integrated-phase-3-safety-results-of-sirukumab-an-anti-il-6-cytokine-monoclonal-antibody-in-patients-with-active-rheumatoid-arthritis/. Retrieved 27 May 2018.
Withdrawal assessment report for Plivensia™ (sirukumab), 14 September 2017. http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2018/02/WC500243181.pdf. Retrieved 29 May 2018.
Ogdie A, Haynes K, Troxel AB, et al. Risk of mortality in patients with psoriatic arthritis, rheumatoid arthritis and psoriasis: a longitudinal cohort study. Ann Rheum Dis. 2014;73:149–53.
Sparks JA, Chang SC, Liao KP, et al. Rheumatoid arthritis and mortality among women during 36 years of prospective follow-up: results from the Nurses’ Health Study. Arthritis Care Res (Hoboken). 2016;68:753–62.
Zhang Y, Lu N, Peloquin C, et al. Improved survival in rheumatoid arthritis: a general population-based cohort study. Ann Rheum Dis. 2017;76:408–13.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
GRB has received grants from Roche, including travel support, and has received consulting fees/payment for lectures from Roche, Sanofi, and Janssen. EF has received grants, consulting/speaker fees from Roche and Sanofi. ABA has received honoraria for lecturing from MSD.
Funding
No sources of funding were used to support the writing of this manuscript.
Rights and permissions
About this article

Cite this article
Avci, A.B., Feist, E. & Burmester, G.R. Targeting IL-6 or IL-6 Receptor in Rheumatoid Arthritis: What’s the Difference?. BioDrugs 32, 531–546 (2018). https://doi.org/10.1007/s40259-018-0320-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-018-0320-3