
Abstract
CT-P13 (Remsima™; Inflectra™), a biosimilar of reference infliximab (Remicade®), is approved by the European Medicines Agency for use in all indications for which reference infliximab is approved, including rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, psoriatic arthritis and psoriasis. Infliximab is a chimeric human-murine monoclonal antibody against the proinflammatory cytokine tumour necrosis factor-α. The CT-P13 infliximab formulation is identical to that of reference infliximab and it has similar physiochemical characteristics. The approval of CT-P13 was based on the results of a rigorous, comparability exercise. This article reviews the results of that exercise, focusing on the clinical evaluation programme. In two well-designed clinical trials, CT-P13 was equivalent to reference infliximab in terms of pharmacokinetic properties in patients with ankylosing spondylitis and in terms of efficacy in patients with rheumatoid arthritis. In both studies, CT-P13 was generally well tolerated with a similar tolerability profile to that of reference infliximab. Immunogenicity evaluations demonstrated that the proportion of patients developing anti-drug antibodies was similar with each agent. Preliminary data from trial extensions demonstrated that in patients who switched from reference infliximab to CT-P13, efficacy was sustained and similar to those who were treated continuously with CT-P13. As with all biosimilar and generic agents, CT-P13 has the potential to reduce treatment costs compared with those of reference infliximab, and modelled analyses predict significant cost savings compared with reference infliximab. In conclusion, CT-P13 is an infliximab biosimilar that provides a useful alternative to reference infliximab in patients requiring infliximab therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The first biosimilar monoclonal antibody approved by the European Medicines Agency. |
Demonstrated similarity to reference infliximab in an extensive comparability exercise, including bioanalytical, preclinical and clinical analyses. |
Approved for use in all indications in which reference infliximab is approved. |
Demonstrated an equivalent pharmacokinetic profile to that of reference infliximab in ankylosing spondylitis patients. |
Demonstrated equivalent efficacy to that of reference infliximab in rheumatoid arthritis patients. |
Generally well tolerated, with a similar tolerability profile to that of reference infliximab. |
The immunological response to CT-P13 was similar to that of reference infliximab. |
1 Introduction
CT-P13 (Remsima™; Inflectra™) is an infliximab biosimilar. A biosimilar is a biotherapeutic agent that is similar to an already licensed reference biotherapeutic agent in terms of quality, safety and efficacy [1]. Unlike generic medicines, which are chemical, small molecule drugs that are structurally and therapeutically equivalent to the reference agent, biotherapeutics consist of relatively large, complex proteins that are more difficult to replicate [1]. As a result, biosimilars must undergo comprehensive and rigorous nonclinical and clinical evaluation in order to demonstrate similarity to the reference biological medicine [1, 2]. The European Medicines Agency (EMA) stipulates that nonclinical evaluation include pharmaco-toxicological analysis, and the clinical evaluation programme include pharmacokinetic, pharmacodynamic (if feasible) and efficacy studies, as well as clinical safety studies with a particular emphasis on the immunogenicity of the biosimilar [2].
The CT-P13 infliximab formulation was developed to closely replicate reference infliximab (Remicade®). It is identical to the pharmaceutical form, composition and strength of reference infliximab, with the same route of administration [3]. CT-P13 received approval for the same therapeutic indications as reference infliximab in September 2013, including rheumatoid arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, psoriatic arthritis and psoriasis. These indications are chronic inflammatory autoimmune disorders characterized by tumour necrosis factor-α (TNF-α)-mediated inflammation. This article reviews the results of the comparability exercise that was required to demonstrate the biosimilarity of CT-P13 to reference infliximab, on which EMA approval of CT-P13 was based, with a focus on the clinical evaluation programme.
2 Product Description and Nonclinical Evaluation
Infliximab is a chimeric human-murine monoclonal antibody (mAb) against the proinflammatory cytokine TNF-α [3]. The drug binds with high affinity to the soluble and transmembrane forms of TNF-α, thereby neutralizing the biological activity of TNF-α [3]. Since the approval of reference infliximab in the EU in 1999, the pharmacodynamic properties of the drug have been well described [4, 5].
The CT-P13 infliximab formulation is identical to that of reference infliximab [3]. In extensive product characterization exercises, CT-P13 was found to be similar to reference infliximab in terms of primary, secondary and higher order structure, including protein folding, and post-translational glycosylation [3].
A series of qualitative and quantitative formulation studies demonstrated that the CT-P13 formulation is adequately robust in terms of product stability and quality, and is similar in this regard to the reference infliximab formulation [3].
In in vitro studies, CT-P13 and reference infliximab demonstrated very similar binding affinities for soluble monometric and trimetric forms of TNF-α and transmembrane TNF-α [3]. The equilibrium dissociation constants (KD) for soluble monometric and trimetric TNF-α were almost identical with both infliximab agents [3]. CT-P13 and reference infliximab also demonstrated similar binding affinities for the Fcγ receptors FcγRI, FcγRIIa and FcRN. A difference in the relative binding affinities for FcγRIIIa was identified, but after further analysis using serum from Crohn’s disease patients, it was determined that the difference would be unlikely to impact biological activity and, therefore, would be unlikely to have any clinical relevance for the efficacy and safety of CT-P13 [3].
Pharmacokinetic analyses comparing CT-P13 and reference infliximab and repeat-dose toxicity studies of intravenous CT-P13 were performed in rats [3]. Overall, the pharmacokinetics of CT-P13 and reference infliximab at doses of 10 and 50 mg/kg in rats were similar. There were no toxicity concerns with CT-P13 in off-target toxicity evaluations [3].
3 Clinical Evaluation
The clinical trial programme to demonstrate biosimilarity between CT-P13 and reference infliximab consisted of a phase 1 pharmacokinetic study in patients with ankylosing spondylitis (Sect. 3.1) [6], and a phase 3 study primarily evaluating efficacy in patients with rheumatoid arthritis (Sect. 3.2) [7]. Both were randomized, double-blind, multinational, parallel-group trials [6, 7]. Results up to 30 weeks are fully published [6, 7], whereas data up to 54 weeks are available as abstracts [8–10]. Results of subsequent open-label, 48-week extension phases, in which patients receiving reference infliximab were switched to CT-P13 are also available as abstracts [11, 12].
In the phase 1 pharmacokinetic study, patients with active ankylosing spondylitis for ≥3 months prior to screening, a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of ≥4 and a visual analog scale score for spinal pain of ≥4 were eligible for randomization to 5 mg/kg of CT-P13 or reference infliximab (see Table 1 for treatment regimen details) [6].
Pharmacokinetic equivalence at steady state (after at least 5 doses of study drug) between CT-P13 and reference infliximab was the primary endpoint, as measured by area under the concentration-time curve (AUC) and the observed maximum serum concentration (Cmax) [6]. Equivalence was demonstrated if the 90 % confidence interval (CI) for the ratio of the mean of each agent was inside the margin of 80–125 %.
The phase 3, efficacy trial recruited patients with active rheumatoid arthritis for ≥1 year prior to screening despite methotrexate therapy for ≥3 months [7]. Participants were required to have ≥6 swollen and ≥6 tender joints and at least two of the following: morning stiffness lasting ≥45 min, serum C reactive protein (CRP) concentration >2.0 mg/dL and erythrocyte sedimentation rate (ESR) >28 mm/h. Oral corticosteroids and non-steroidal anti-inflammatory drugs (NSAIDs) were permitted if doses had been stable for ≥4 weeks prior to screening. Patients were randomized to 3 mg/kg CT-P13 or reference infliximab (see Table 3 for treatment regimen details).
The primary endpoint in the phase 3 trial was to demonstrate equivalent efficacy with CT-P13 and reference infliximab, as determined by the American College of Rheumatology 20 % (ACR20) response at week 30 in the intent-to-treat (ITT) population [7]. Equivalence was demonstrated if the 95 % CI for treatment difference was within ±15 %, based on historical clinical trial data.
3.1 Pharmacokinetic Properties
The pharmacokinetics (AUC and Cmax) of CT-P13 were equivalent to those of reference infliximab, with the ratio of geometric means close to 100 % for both AUC and Cmax at steady state in patients with ankylosing spondylitis (Table 1) [6]. In the subgroup of patients who were anti-drug antibody negative, AUC and Cmax geometric means in the two treatment groups were numerically greater than in the total population, but the ratio of geometric means remained close to 100 % (Table 1).
With regard to secondary pharmacokinetic endpoints, values were also similar between CT-P13 and reference infliximab at steady state (between weeks 22 and 30) (Table 2) [6].
Pharmacokinetic parameters continued to be similar with CT-P13 and reference infliximab during 8-weekly administration up to 54 weeks in 213 ankylosing spondylitis patients [8]. The 90 % CI for the ratio of geometric means continued to be inside the margin of 80–125 %. As demonstrated at 30 weeks, at week 54 patients who were anti-drug antibody negative had numerically greater AUC and Cmax geometric means than those who were anti-drug antibody positive [8].
In patients with rheumatoid arthritis currently receiving methotrexate, values of pharmacokinetic endpoints after each infusion throughout 30 weeks of treatment were very similar for CT-P13 and reference infliximab (Table 2) [7]. Geometric mean Cmax values for doses 1–6 ranged from 83.9 to 111.9 µg/mL for CT-P13 and 83.8 to 105.1 µg/mL for reference infliximab [7]. In the subgroup of patients who were anti-drug antibody negative, geometric mean Cmax and Cmin values at steady-state in both treatment groups were numerically greater than those recorded in anti-drug antibody positive patients (i.e. ≈48 % of patients at week 30).
3.2 Efficacy
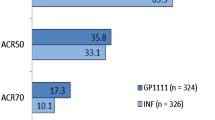
The efficacy of CT-P13 was equivalent to that of reference infliximab, with the CI for the treatment difference for ACR20 response at week 30 falling well within ±15 % in patients with rheumatoid arthritis receiving methotrexate (ITT population) (Table 3) [7]. Similarly, results for ACR50 (Table 3) and ACR70 response analyses at weeks 14 and 30 in the ITT and per-protocol (PP) populations were very similar in the two treatment groups. At week 30, the ACR70 response in the CT-P13 and reference infliximab groups was 20.2 and 17.9 %, respectively, in the PP population equating to a treatment difference of 2 % (95 % CI −5 to 9 %) [7]. In a post-hoc analysis of ACR20 response rates based on baseline CRP levels, similar results were demonstrated in the CT-P13 and reference infliximab groups among those with CRP >2 mg/dL (58.7 and 58.6 %, respectively) or ≤2 mg/dL (61.9 and 58.5 %).
All additional secondary endpoints in the rheumatoid arthritis study demonstrated similar results with CT-P13 and reference infliximab (most are summarized in Table 4) [7]. Good or moderate European League Against Rheumatism (EULAR) responses (CRP) at week 30 were recorded in 85.8 % of CT-P13 recipients and 87.1 % of reference infliximab recipients [relative risk (RR) 0.98; 95 % CI 0.92–1.06]. The mean improvement in CRP levels was −0.6 and −0.8 mg/dL, respectively, at week 14, and these values were unchanged at week 30. The proportion of patients in CT-P13 and reference infliximab groups requiring salvage therapy [e.g. NSAID or disease-modifying antirheumatic drugs (DMARDs)] throughout 30 weeks of treatment was 3.2 and 4.0 %, respectively, and the median time to the onset of an ACR20 response was 99 and 100 days [7].
Among 457 rheumatoid arthritis patients who continued 8 weekly therapy for 54 weeks, ACR20, ACR50 and ACR70 response rates continued to be very similar in groups receiving CT-P13 or reference infliximab [9]. For example, an ACR20 response was achieved in 57.0 and 52.0 % of patients (ITT analysis) in each group, respectively (95 % CI for treatment difference −0.03 to 0.13) [9]. In a health-related quality of life (HR-QOL) assessment carried out at week 54, there were no significant differences between groups receiving CT-P13 or reference infliximab in the Health Assessment Questionnaire (HAQ) disability index or the Short-Form 36 (SF-36), with both treatments leading to improvements of a similar magnitude that were sustained over the treatment period [10].
In the phase 1 study in patients with ankylosing spondylitis (n = 250), efficacy was also very similar between groups receiving CT-P13 or reference infliximab [6]. A 20 % response in the Assessment in Ankylosing Spondylitis (ASAS20) criteria was reported in 62.6 % of CT-P13 recipients and 64.8 % of reference infliximab recipients at 14 weeks (OR 0.91; 95 % CI 0.53–1.54), and values in corresponding groups at week 30 were 70.5 and 72.4 % (OR 0.91; 95 % CI 0.51–1.62). An ASAS40 response in each of the respective groups was reported in 41.7 and 45.9 % of patients at 14 weeks (OR 0.85; 95 % CI 0.51–1.42), and 51.8 and 47.4 % at 30 weeks (OR 1.19; 95 % CI 0.70–2.00) [6].
All other efficacy evaluations in the phase 1 study demonstrated a similar treatment response with CT-P13 and reference infliximab, including the BASDAI, Bath Ankylosing Spondylitis Functional Index, Bath Ankylosing Spondylitis Metrology Index, chest expansion score, and the Ankylosing Spondylitis Disease Activity Score assessed according to baseline CRP levels [6]. HR-QOL improved significantly from baseline and to a similar extent in both treatment groups, as measured by the SF-36, in which the physical component improved from baseline to week 30 in the CT-P13 and reference infliximab groups by a median of 7.6 and 8.5, respectively [6].
Among 213 ankylosing spondylitis patients who continued 8-weekly therapy for 54 weeks, response rates continued to be similar in CT-P13 and reference infliximab groups [8]. For example, an ASAS40 response was achieved in 54.7 and 49.1 % of patients in each group, respectively.
In both studies, the efficacy response in both treatment groups was less robust in anti-drug antibody positive patients than in anti-drug antibody negative patients throughout 54 weeks of treatment [6–9]. There were no statistically significant differences between treatment groups in efficacy analyses at 30 weeks based on anti-drug antibody status in either trial [6, 7].
3.2.1 Switching from Reference Infliximab to CT-P13
In the open-label, 48-week extension phases of both studies, efficacy was sustained and similar in patients who continued CT-P13 treatment and those who switched from reference infliximab to CT-P13 at the end of the first year [11, 12]. At week 102 in the phase 3 study in rheumatoid arthritis patients (n = 302), ACR20 response rates were 72.2 and 71.8 % in each group, respectively [12]. At week 102 in the phase 1 study in ankylosing spondylitis patients (n = 174), ASAS40 response rates were 63.9 and 61.5 % in each group, respectively [11].
3.3 Immunogenicity
The detection of anti-drug antibodies was similar in CT-P13 and reference infliximab treatment groups in both trials [6, 7]. The proportion of anti-drug antibody positive patients did not increase markedly after 30 weeks of treatment.
Among patients with ankylosing spondylitis, the proportion with antibodies to infliximab in the CT-P13 and reference infliximab groups was 9.1 % (n = 11) and 11.0 % (n = 13), respectively, at week 14, 27.4 % (n = 32) and 22.5 % (n = 25) at week 30 [6], and 22.9 % (n = 25/109) and 26.7 % (n = 28/105) at week 54 [8]. In the extension phase (n = 174), the proportion of patients who were anti-drug antibody positive at week 102 was similar in those who continued CT-P13 therapy and those who were switched from reference infliximab to CT-P13 at the end of the first year (25.0 and 30.7 %, respectively) [11].
Among patients with rheumatoid arthritis, the proportion with antibodies to infliximab in the CT-P13 and reference infliximab groups was 25.4 % (n = 69) and 25.8 % (n = 70), respectively, at week 14, 48.4 % (n = 122) and 48.2 % (n = 122) at week 30 [7], and 52.3 and 49.5 % at week 54 [9]. In the extension phase (n = 302), the proportion of patients who were anti-drug antibody positive at 2 years was similar in those who continued CT-P13 therapy and those who were switched from reference infliximab to CT-P13 at the end of the first year (46.4 and 49.6 %, respectively) [12].
3.4 Safety
Overall, across both trials, the safety profile of CT-P13 was similar to that of reference infliximab [6, 7]. The majority of adverse events were mild to moderate in severity.
Throughout 30 weeks, treatment-emergent adverse events were reported in 64.8 % of CT-P13 recipients and 63.9 % of reference infliximab recipients in the ankylosing spondylitis trial (n = 250) [6], and 60.1 and 60.8 %, respectively, in the rheumatoid arthritis trial (n = 606) [7]. The most commonly reported treatment-related adverse events were similar in both trials. In patients with rheumatoid arthritis, the most commonly reported treatment-related adverse events in CT-P13 and reference infliximab groups were latent tuberculosis [TB; i.e. patients who were initially negative according to the interferon γ-release assay using Quantiferon-TB Gold in tube (QTF-TB-GIT) but became positive, and didn’t have any clinical symptom or sign of active TB; 4.3 and 4.7 %, respectively], raised ALT (4.0 and 3.7 %), raised AST (2.7 and 2.7 %), urinary tract infection (1.3 and 2.3 %), flare in rheumatoid arthritis activity (2.3 and 1.3 %), nasopharyngitis (2.0 and 1.3 %) and headache (1.3 and 2.0 %) [7]. Infusion-related reactions occurred in 6.6 % of patients in the CT-P13 group and 8.3 % of patients in the reference infliximab group.
Serious treatment-related adverse events among patients with rheumatoid arthritis occurred in 10.0 % of patients receiving CT-P13 and 7.0 % of patients receiving reference infliximab [7]. Three cases of active TB occurred in the CT-P13 group (one in the Philippines, Poland and Mexico) and none in the reference infliximab group. Patients with latent TB who received prophylactic TB medication did not convert to active TB. Two patients in the reference infliximab group withdrew from the trial because of malignancy (breast and cervical) [7]. In patients with ankylosing spondylitis, serious treatment-related adverse events occurred in 4.7 % of patients receiving CT-P13 and 6.4 % of patients receiving reference infliximab [6]. There were no deaths in either treatment group of both trials [6, 7].
3.4.1 Switching from Reference Infliximab to CT-P13
In the open-label, 48-week extension of the phase 3, rheumatoid arthritis trial (n = 302) (total treatment period 102 weeks), the proportion of patients with at least 1 treatment-emergent adverse event in the group maintained on CT-P13 and the group who switched from reference infliximab to CT-P13 was similar (53.5 and 53.8 %, respectively) [12]. Severe treatment-emergent adverse events occurred in 7 (4.4 %) and 8 (5.6 %) patients, respectively, and serious adverse events occurred in 12 (7.5 %) and 13 (9.1 %) patients [12]. Among patients who continued CT-P13 or switched from reference infliximab to CT-P13 in the 48-week extension of the phase 1, ankylosing spondylitis trial (n = 174), the incidence of severe [3 (3.3 %) and 5 (6.0 %) patients, respectively] and serious [4 (4.4 %) and 4 (4.8 %) patients] treatment-emergent adverse events was generally consistent with that reported in the phase 3 trial [11].
4 Pharmacoeconomic Considerations
Because reference infliximab and similar agents have a relatively high purchase price, it is hoped that the introduction of biosimilars will lessen the burden on healthcare budgets. It is also hoped that the improved affordability of these agents will lead to improved access, particularly in lower income European countries, where patients have less access than those in higher income countries [13, 14].
Two recent modelled, budget impact analyses estimated that introducing CT-P13 as a treatment option for patients with rheumatoid arthritis in six central European countries (Bulgaria, Czech Republic, Hungary, Poland, Romania and Slovakia) [15] or in Ireland [16] could result in significant cost savings (results are reported in abstracts and therefore provide limited methodological details).
The analyses were modelled from the perspective of a third party payer over a 3- [15] or a 5-year [16] time horizon and estimated that the price of CT-P13 would be 25 [15] or 20 % [16] lower than that of reference infliximab. One model estimated that if 80 % of rheumatoid arthritis patients in six central European countries were gradually switched from reference infliximab to CT-P13, a net benefit over 3 years of €29,810,000 would result compared to the scenario of CT-P13 not being available for use [15]. The Irish model predicted a cumulative cost saving over 5 years of up to €5,313,184 if all existing and new patients eligible for infliximab commenced CT-P13 in the first year [16].
As with all modelled pharmacoeconomic analyses, these analyses are subject to limitations, with the potential for input data to differ from real-life situations.
5 Dosage and Administration
Dosage and administration details for CT-P13 are the same as those for reference infliximab. CT-P13 should be administered as an intravenous infusion over 2 h; shortened infusion times (no less than 1 h) may be considered in selected patients who have tolerated at least three initial 2-h infusions [17, 18]. Patients should be observed for infusion-related reactions for at least 1–2 h after the infusion. Emergency equipment must be available, including adrenaline, antihistamines, corticosteroids and an artificial airway. Pretreatment with agents such as an antihistamine or hydrocortisone may be considered [17, 18]. See Table 5 for a summary of treatment regimen details in various indications.
As TNF-blockers are associated with an increased susceptibility to serious infections, patients receiving infliximab should be monitored closely for bacterial infections (including TB, sepsis and pneumonia) and invasive fungal and viral infections (including hepatitis B) [17, 18]. The risk of infection is greater in the elderly (aged ≥65 years) and in children than in adults, and care should be taken with infliximab in these populations. The risk of developing lymphomas or other malignancies in patients treated with TNF-blocking agents cannot be ruled out, and caution should be used when infliximab is administered in patients with a history of malignancy or who develop a malignancy [17, 18]. Local prescribing information should be consulted for comprehensive information regarding dosage, warnings, precautions and contraindications.
6 Current Status of CT-P13
CT-P13 is the first biosimilar monoclonal antibody to be approved by the EMA [3]. Such agents have been developed in recent years as the first biological therapies approach patent expiry; the patent for reference infliximab will expire in the EU in August 2014. Due to the complexities of replicating reference biological agents (summarized by Dorner et al. [19]), a biosimilar must undergo extensive, and highly regulated (by the EMA [2] and US FDA [20]) comparability exercises, which include demonstrating similar pharmacokinetics, efficacy and safety to the reference agent in well designed, randomized, controlled trials.
The approval of CT-P13 was based on the results of two pivotal clinical trials, in which the pharmacokinetics (in patients with ankylosing spondylitis; Sect. 3.1) and efficacy (in patients with rheumatoid arthritis; Sect. 3.2) of CT-P13 were equivalent to reference infliximab. Clinical trials also demonstrated that CT-P13 has a similar tolerability profile to that of reference infliximab (Sect. 3.4), and immunogenicity evaluations demonstrated that the proportion of patients developing anti-drug antibodies was similar with each agent (Sect. 3.3). Preliminary data from trial extensions demonstrated that in patients who switched from reference infliximab to CT-P13, efficacy was sustained and similar to those who were maintained on CT-P13 (Sect. 3.2.1). As required by regulatory authorities, post authorization studies and registries will provide further valuable long-term efficacy and safety data on CT-P13. In particular, serious infections, including TB, will be closely monitored over the long term and in a larger population than that included in clinical trials to date.
The overall comparability testing of CT-P13 to reference infliximab, including bioanalytical and preclinical analyses, and clinical data from the two comparative trials in rheumatology disorders, has enabled extrapolation of CT-P13 approval to all other indications for which reference infliximab is approved, including Crohn’s disease, ulcerative colitis, psoriatic arthritis and psoriasis (Table 5) [3]. According to preliminary data based on modelled analyses, if the price of CT-P13 is markedly lower than that of reference infliximab, the use of CT-P13 instead of reference infliximab is likely to lead to cost savings (Sect. 4).
In conclusion, CT-P13 is an infliximab biosimilar with proven pharmacokinetic and therapeutic equivalence to reference infliximab and, thus, provides a useful alternative in patients requiring infliximab therapy.
Data selection sources:
Relevant medical literature (including published and unpublished data) on CT-P13 was identified by searching databases including MEDLINE (from 1946) and EMBASE (from 1996) [searches last updated 28 March 2014], bibliographies from published literature, clinical trial registries/databases and websites. Additional information was also requested from the company developing the drug.
Search terms: CT-P13, biosimilar, infliximab, Remsima, Inflectra
Study selection: Studies in patients who received CT-P13. When available, large, well designed, comparative trials with appropriate statistical methodology were preferred.
References
World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs); 2009. http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed 19 Dec 2013.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Draft; 2013. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/06/WC500144124.pdf. Accessed 17 Feb 2014.
European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Assessment report: Inflectra (infliximab); 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002778/WC500151490.pdf. Accessed 17 Feb 2014.
Siddiqui MAA, Scott LJ. Infliximab: a review of its use in Crohn’s disease and rheumatoid arthritis. Drugs. 2005;65:2179–208.
Keating GM, Perry CM. Infliximab: an updated review of its use in Crohn’s disease and rheumatoid arthritis. BioDrugs. 2002;16(2):111–48.
Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–12.
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–20.
Park W, Jaworski J, Brzezicki J, et al. A randomised, double-blind, parallel-group, phase 1 study comparing the pharmacokinetics, safety and efficacy of CT-P13 and infliximab in patients with active ankylosing spondylitis: 54 week results from the PLANETAS study [abstract no. FRI0421]. 14th Annual Congress of the European League Against Rheumatism, Madrid; 12–15 Jun 2013.
Yoo DH, Racewicz A, Brzezicki J, et al. A phase 3 randomised controlled trial to compare CT-P13 with infliximab in patients with active rheumatoid arthritis: 54 week results from the PLANETRA study [abstract no. OP0068]. 14th Annual Congress of the European League Against Rheumatism, Madrid; 12–15 Jun 2013.
Yoo D, Yagensky A, Toncheva A, et al. Impact of CT-P13 and originator infliximab treatment on quality of life derived from the Health Assessment Questionnaire (AQ) and Short-Form 36 (SF-36) from a randomized, double-blind trial in patients with active RA [abstract no. 2392]. ACR/ARHP Annual Meeting, San Diego; 26–30 Oct 2013.
Park W, Miranda P, Brzosko M, et al. Efficacy and safety of CT-P13 (infliximab biosimilar) over two years in patients with ankylosing spondylitis: comparison between continuing with CT-P13 and switching from infliximab to CT-P13 [abstract no. L15]. ACR/ARHP Annual Meeting, San Diego; 26–30 Oct 2013.
Yoo DH, Prodanovic N, Jaworski J, et al. Efficacy and safety of CT-P13 (infliximab biosimilar) over two years in patients with rheumatoid arthritis: comparison between continued CT-P13 and switching from infliximab to CT-P13 [abstract no. L1]. ACR-ARHP Annual Meeting, San Diego; 26–30 Oct 2013.
Putrik P, Ramiro S, Kvien TK, et al. Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis. 2014;73:198–206.
Putrik P, Ramiro S, Kvien TK, et al. Variations in criteria regulating treatment with reimbursed biologic DMARDs across European countries. Are differences related to country’s wealth? Ann Rheum Dis. 2013. doi:10.1136/annrheumdis-2013-203819.
Brodszky V, Pentek M, Baji P, et al. Budget impact analysis of biosimilar infliximab treatment for rheumatoid arthritis in six Central European countries [abstract no. PMS21]. Value Health. 2013;16(7):A558.
McCarthy G, Ebel Bitoun C, Guy H. Introduction of an infliximab biosimilar (CT-P13): a five-year budget impact analysis for the treatment of rheumatoid arthritis in Ireland [abstract no. PMS22]. Value Health. 2013;16(7):A558.
European Medicines Agency. Remsima: summary of product characteristics; 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002576/WC500150871.pdf. Accessed 19 Dec 2013.
European Medicines Agency. Inflectra: summary of product characteristics; 2013. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002778/WC500151489.pdf. Accessed 19 Dec 2013.
Dorner T, Strand V, Castaneda-Hernandez G, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis. 2013;72(3):322–8.
US Food and Drug Administration. Quality considerations in demonstrating biosimilarity to a reference protein product: draft guidance; 2012. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291134.pdf. Accessed 13 Jan 2014.
Lipsky PE, van der Heijde DM, St Clair EW, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343(22):1594–602.
St Clair EW, Van der Heijde DM, Smolen JS, et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis Rheum. 2004;50(11):3432–43.
Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–9.
Sands BE, Anderson FH, Bernstein CN, et al. Infliximab maintenance therapy for fistulizing Crohn’s disease. N Engl J Med. 2004;350:876–85.
Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462–76.
van der Heijde D, Dijkmans B, Geusens P, et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis: results of a randomized, placebo-controlled trial (ASSERT). Arthritis Rheum. 2005;52(2):582–91.
Antoni CE, Kavanaugh A, Kirkham B, et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheum. 2005;52(4):1227–36.
Antoni CE, Krueger GG, de Vlam K, et al. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann Rheum Dis. 2005;64:1150–7.
Gottlieb AB, Evans R, Li S, et al. Infliximab induction therapy for patients with severe plaque-type psoriasis: a randomized, double-blind, placebo-controlled trial. J Am Acad Dermatol. 2004;51(4):534–42.
Reich K, Nestle FO, Papp K, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366(9494):1367–74.
Hyams J, Crandall W, Kugathasan S, et al. Induction and maintenance infliximab therapy for the treatment of moderate-to-severe Crohn’s disease in children. Gastroenterology. 2007;132(3):863–73.
Hyams J, Damaraju L, Blank M, et al. Induction and maintenance therapy with infliximab for children with moderate to severe ulcerative colitis. Clin Gastroenterol Hepatol. 2012;10(4):391–9.
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process, the manufacturer of the agent under review was offered an opportunity to comment on this article. Changes resulting from comments received were made by the author on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Additional information
The manuscript was reviewed by: M. T. Nurmohamed, Rheumatology and Internal Medicine, VU University Medical Center & Reade, Amsterdam Rheumatology Immunology Center, Amsterdam, The Netherlands; D. L. Scott, Department of Rheumatology, Kings College London School of Medicine, London, UK; D. H. Yoo, Division of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Department of Internal Medicine Hanyang University College of Medicine, Seoul, Republic of Korea.
Rights and permissions
About this article
Cite this article
McKeage, K. A Review of CT-P13: An Infliximab Biosimilar. BioDrugs 28, 313–321 (2014). https://doi.org/10.1007/s40259-014-0094-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-014-0094-1