
Abstract
Fibrotic diseases such as idiopathic pulmonary fibrosis or scleroderma (systemic sclerosis) are chronic fibroproliferative disorders for which there are currently no effective treatments. Dysregulated normal tissue repair process is considered to cause a fibrotic response culminating in compromised organ function due to excess extracellular matrix deposition. The mechanisms underlying the pathophysiology of fibrosis are poorly understood. Recent findings suggest that focal adhesion kinase (FAK) plays a key role in development of fibrotic disorders, and it appears to be an attractive target for antifibrotic therapy. Here, we review the emerging role of FAK as a key regulator of fibrotic signaling and its potential as a future therapeutic target to counteract fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Chronic fibrotic disorders, including idiopathic pulmonary fibrosis (IPF) or systemic sclerosis (SSc), contribute towards significant mortality and morbidity worldwide [1, 2]. Unfortunately, these diseases are unresponsive to the majority of currently available pharmacologic therapies [2]. Fibrogenesis is considered as the result of a dysregulated wound healing response where the activation of fibroblasts into alpha-smooth muscle actin (α-SMA)–positive myofibroblasts is an essential step in the evolution of fibrotic disorders. Myofibroblasts are responsible for the production and deposition of the extracellular matrix (ECM) components that are a hallmark of the disease, which leads to the destruction of organ architecture [3, 4]. The profibrotic factor transforming growth factor beta-1 (TGF-β1) has been shown to be an important mediator of tissue fibrosis and can induce myofibroblast formation in vitro and in vivo [5–9]. Recent work during the past 10 years has also focused interest on many other factors that fine tune myofibroblast differentiation, including the ED-A splice variant of fibronectin, endothelial-derived factors such as endothelin-1 (ET-1), the matricellular proteins, which include connective tissue growth factor (CCN2/CTGF) or variations in matrix stiffness [10–13]. These factors influence fibroblast adhesion to the matrix, typically through integrin binding and activation of focal adhesion kinase (FAK). In fact, it is now appreciated that increased adhesive signaling and FAK activation is a hallmark of lesional fibroblasts [14]. To date, the role for FAK in tissue fibrogenesis is just beginning to unravel, although a few studies have recently explored the role of FAK in lung and skin biology. The mechanisms leading to myofibroblast differentiation are not fully understood, and studies on the factors and signaling pathways that govern myofibroblast formation will be necessary for the design of new therapeutic strategies aimed at counteracting fibrotic disorders. In this review, we integrate recent findings regarding the emerging role of FAK in myofibroblast differentiation and fibrotic disorders such as IPF or SSc.
2 Focal Adhesion Kinase (FAK)
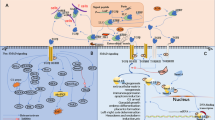
FAK is a 125-kDa non-receptor cytoplasmic tyrosine kinase that was firstly identified in 1992 as a highly tyrosine-phosphorylated protein that resides at sites of integrin clustering—the so-called focal adhesions [15–17]. Today FAK is well recognized as an important mediator of both integrin and growth-factor signaling [18]. FAK is a major regulator of cell proliferation, differentiation, survival and migration [19]. FAK is a ubiquitously expressed protein that is composed of an N-terminal FERM (protein 4.1, ezrin, radixin and moesin sequence homology) domain, a central kinase domain, 3 proline-rich regions and a C-terminal focal-adhesion targeting (FAT) domain (Fig. 1) [20]. The FERM domain binds to sequences in the cytoplasmic domain of β-integrin subunits targeting FAK to sites of integrin or growth factor receptor clustering and facilitates a signaling linkage from receptor tyrosine kinases (RTK) [21–23]. The FAT region at the C-terminal domain of FAK promotes the colocalization of FAK with integrins at focal contacts [24]. The C-terminal, non-catalytic domain of FAK, termed FRNK (FAK-related-non-kinase), is expressed independently in certain cells and may function as negative endogenous regulator of FAK kinase activity (Fig. 1) [25].

Focal adhesion kinase (FAK) domains and phosphorylation sites. The N-terminal FERM (protein 4.1, ezrin, radixin and moesin sequence homology) domain directs interactions of FAK with epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) receptors as well as integrins. FAK also contains three proline-rich regions (PRR1–3), which bind SH3 domain-containing proteins such as p130Cas (protein 130 kDa Crk-associated substrate), GRAF (GTPase regulator associated with FAK) and ASAP1 (ArfGAP with SH3 domain, ankyrin repeat and PH domain 1). FAK is phosphorylated (P) on different tyrosine residues, including Y397, Y407, Y576, Y577, Y861 and Y925. FAK phosphorylation on Y397 results in the binding of SH2 domain-containing proteins including sarcoma (Src), phospholipase C gamma (PLCγ) and phosphatidylinositol 3-kinase (PI3 K) among other proteins. Phosphorylation of Y576 and Y577 within the kinase domain drives FAK to full kinase activity. FAK phosphorylation at Y925 creates a binding site for GRB2 adaptor protein leading to mitogen-activated protein kinase (MAPK) cascade activation. The C-terminal domain encompasses the focal-adhesion targeting (FAT) domain that targets FAK to focal adhesions by associating with proteins such as talin and paxillin. The FAT domain also links FAK to the activation of Ras homolog gene family (Rho) GTPases by binding to p190 Rho guanine nucleotide exchange factor (RhoGEF). FAK-related non-kinase (FRNK) is autonomously expressed and has an identical sequence to the C-terminal domain of FAK. GTP guanosine triphosphate
FAK activation is primarily mediated by autophosphorylation of FAK on Y397 that occurs in response to many stimuli, including environmental cues and soluble growth factor signaling through RTK or G protein coupled receptors (GPCRs) [26, 27]. Phosphorylation of FAK on Y397 results in a high affinity binding site for Src (sarcoma) kinase, thereby creating a functional bipartite kinase complex (Fig. 1) [28]. The association of Src with FAK results in the activation of the kinase activity of Src. Now, Src phosphorylates FAK on Y576 and Y577 within the FAK catalytic domain and leads to the full enzymatic activity of FAK [29]. Src can further phosphorylate FAK on Y407, Y861, and Y925 with phosphorylated Y925 acting as a docking site for growth-factor-receptor bound protein 2 (GRB2), which results in the activation of the RTK/Ras guanosine triphosphate (GTP)ase/mitogen-activated protein (MAP) kinase (MAPK) cascade (Fig. 1). Functionally, the FAK/Src complex regulates cell adhesion, motility, and migration, as well as cell growth and survival.
3 Role of FAK in Fibrosis
In the past two decades, extensive research has been performed to investigate the role of FAK during embryonic development and in the pathogenesis of human disease, including the progression of multiple mesenchymal and epithelial malignant tumors, Alzheimer’s, rheumatoid arthritis, cardiac hypertrophy, hypertension, and atherosclerosis [30–35]; in addition, its role in fibrotic disorders such as SSc or IPF is now emerging. FAK has attracted special attention for its role in mediating fibrotic responses, and that its inhibition could be of therapeutic importance in counteracting profibrotic mechanisms. Recent in vitro and in vivo studies document the role of FAK in fibrotic signaling in mice and humans, although much remains to be explored regarding the mechanisms by which FAK contributes to fibrogenesis.
3.1 Role of FAK in Myofibroblast Differentiation
FAK plays a central role in mediating adhesive signaling through integrin activation but also participates in transduction pathways activated by growth factors via GPCRs and RTKs (Fig. 2). Emerging in vitro data has focused interest on the regulatory mechanisms by which FAK contributes to myofibroblast formation in fibrotic diseases.

Focal adhesion kinase (FAK) integrates growth factor and integrin signals to promote myofibroblast formation and fibrosis. FAK has been shown to be important in transducing mechanical stimuli to downstream biochemical pathways that lead to fibroblast activation. FAK is also involved in transducing signals from growth factors such as transforming growth factor beta-1 (TGF-β1) or platelet-derived growth factor (PDGF) through their specific receptor tyrosine kinases (RTKs). Several bioactive peptides, including endothelin, lysophosphatidic acid (LPA) and Ang II (angiotensin II), also activate FAK via their specific cell-surface G protein coupled receptor (GPCR). Taken together, these factors regulate anoikis resistance phenotype, cell migration, and myofibroblast differentiation in a FAK-dependent manner. TGF-β1 and endothelin-1 (ET-1) cause FAK activation leading to TGF-β-activated kinase 1 (TAK1) and c-Jun N-terminal kinase (JNK) phosphorylation to ultimately promote fibroblast activation. Therefore, migration and survival of alpha-smooth muscle actin (α-SMA)-positive myofibroblast-producing collagen type I are key steps for the development of fibrosis. Peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands inhibit TGF-β1-induced myofibroblast differentiation by targeting the FAK pathway. Thus, FAK functions as a common target of multiple factors including environmental cues and soluble profibrotic factors. In summary, FAK is a central mediator of fibrogenesis and FAK inhibition may represent a useful therapeutic tool in the treatment of fibrotic diseases. AP-1 Activator protein 1, CCN2/CTGF connective tissue growth factor, ECM extracellular matrix, P phosphate group, PI3 K phosphatidylinositol 3-kinase, PKB protein kinase B, Rac Ras-related C3 botulinum toxin substrate, Rho Ras homolog gene family, SMAD3 Smad family member 3: the SMAD proteins are homologs of both the Drosophila protein, mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein SMA (from gene sma for small body size), Src sarcoma, ↑ indicates increase
Myofibroblasts are pathogenic in pulmonary fibrotic disease due to their excessive production of ECM within the lung that typically results in respiratory failure. TGF-β1 is a well known inducer of myofibroblast differentiation in the lung, but the molecular mechanisms for this effect remain obscure. Recent data demonstrated that TGF-β1-induced myofibroblast differentiation of lung fibroblasts is dependent on adhesion-mediated signaling through FAK activation [36]. In this regard, pharmacologic inhibition of FAK or overexpression of kinase-deficient FAK inhibits TGF-β1-induced α-SMA expression [36, 37]. Recent molecular studies showed that JNK (c-Jun N-terminal kinase) and TAK1 (TGF-β-activated kinase 1) kinase operate downstream of FAK/Src in mediating fibrogenic responses upon TGF-β1 stimulation [38, 39]. Proliferation and expansion of interstitial fibroblasts are predominant features of progressive chronic kidney diseases. Thus, it has been shown that MAP-kinase activity necessary for TGF-β1-stimulated type I collagen expression requires FAK activity in human kidney mesangial cells [40]. In rat hepatic stellate cells (HSC), specific shRNA (short hairpin RNA) targeting of FAK attenuated ECM synthesis and promoted ECM degradation, making FAK a potential target for novel anti-fibrosis therapies in hepatic fibrosis [41]. Importantly, FAK has also been implicated in TGF-β1-induced renal tubular epithelial-to-mesenchymal transition (EMT) [42]. In contrast to this line of thinking, another study shows that FAK was not necessary for TGF-β1-mediated myofibroblast differentiation, but FAK was necessary for fibroblast growth factor and heparin (FGF/h)-mediated inhibition of myofibroblast differentiation [43].
ET-1 is widely accepted as one of the main profibrotic factors that cause fibroblast activation [44]. More recently, the present authors have shown that β1-integrin/FAK signaling directs ET-1-induced myofibroblast differentiation [45].
Recent experiments suggest that some endogenous mechanisms operate to downregulate FAK activation in myofibroblasts. Firstly, the non-catalytic FAK-related non-kinase FRNK has been shown to abrogate TGF-β1-induced myofibroblast differentiation in vitro and in vivo [46]. Prostaglandin E2 (PGE2) was also identified to have the potential to limit TGF-β1-induced myofibroblast differentiation by inhibiting FAK activation [47]. Similarly, peroxisome proliferator-activated receptor-γ (PPAR-γ), a key anti-fibrotic factor [48], when activated by its ligands, inhibits TGF-β1-induced profibrotic mechanism by targeting the FAK pathway [49].
3.2 Role of FAK in Cell Survival
Myofibroblast apoptosis is critical for the normal resolution of a wound healing program and impaired myofibroblast apoptosis is associated with tissue fibrosis [50]. FAK deactivation has been associated with apoptosis induced by loss of adhesion (termed ‘anoikis’) [51, 52]. TGF-β1 functions as an anti-apoptotic signal for fibroblasts/myofibroblasts during tissue fibrogenesis. Recently, it has been demonstrated that TGF-β1/SMAD3Footnote 1/FAK signaling promotes anoikis-resistant myofibroblast phenotype [53]. Recent findings support the notion that TGF-β1 promotes an antiapoptotic phenotype in myofibroblasts by the activation of the FAK pathway and the prosurvival protein kinase B/Akt (PKB/Akt) pathway [37]. In this regard, FAK-mediated PKB/Akt activation in fibroblasts is under phosphatidylinositol 3-kinase (PI3K) control [51, 54]. Thus, the pathological FAK/PI3K/Akt signaling pathway enhances proliferation and survival of primary lung fibroblasts [51, 55]. In contrast, it has been recently shown that TGF-β1 co-ordinately and independently activates the FAK and AKT signaling pathways to confer an anoikis-resistant phenotype to myofibroblasts [53]. In a sequential model, PGE2 diminishes TGF-β1-induced phosphorylation of FAK and, in turn, limits activation of PKB/Akt pathway [47]. Moreover, another in vitro study defines a novel mechanism by which ET-1 promotes myofibroblast resistance to apoptosis through FAK-dependent upregulation of survivin [56].
3.3 Role of FAK in Cell Migration
Fibroblast and myofibroblast migration plays an important role in the normal wound healing process; however, increased cell migration may contribute to the accumulation of myofibroblast in the fibrotic lesions, thus contributing to the development of fibrosis [57]. FAK can regulate cell motility by influencing the cytoskeleton organization, structures of focal adhesions and membrane protrusions [18]. To note, FAK-deficient fibroblasts show significantly decreased cell migration, and re-expression of FAK in those cells restores the cell migration [58]. Expression of the mutated Y397F FAK effectively inhibits FAK-mediated cell migration. Previous studies have shown that FAK is activated in response to diverse pro-migratory stimuli including lysophosphatidic acid (LPA) and platelet-derived growth factor (PDGF) [59, 60]. In fact, LPA and PDGF-mediated myofibroblast migration through FAK activation contribute to the accumulation of hepatic myofibroblast and hence to the development of hepatic fibrosis [61]. Interestingly, endogenous FRNK expression is inversely correlated with FAK activation and cell migration rate in lung fibroblasts. Accordingly, FRNK overexpression abrogates cell migration, and blocks the activation of FAK in lung fibroblasts [46].
3.4 FAK in Human Fibrotic Disorders
Recent evidence clearly shows that fibrotic cells often display persistent FAK activation and enhanced adhesion capacity, but regulatory mechanisms for this effect remain elusive. Lesional skin fibroblasts taken from scleroderma patients display increased FAK expression and activity when compared with normal human skin fibroblasts [14]. TGF-β has been shown to activate FAK in human skin fibroblasts from control donors and SSc patients. Accordingly, pharmacologic or genetic FAK inhibition results in the downregulation of TGF-β-induced pro-fibrotic genes including collagen type I and α-SMA expression in fibroblasts [14]. In addition to the research on the skin, studies performed on pulmonary fibrosis provide further evidence of the role of FAK in fibrosis. We have recently shown that FAK expression and activity were upregulated in fibroblast foci and remodeled vessels from lung fibrosis patients [45]. Treatment of lung myofibroblasts with FAK inhibitors in these cells has been shown to abrogate the ability of TGF-β1 or ET-1 to induce myofibroblast differentiation and collagen deposition [36, 45]. Moreover, previous data demonstrate that lung fibroblasts from IPF patients have increased cell migration and FAK phosphorylation when compared with normal human lung fibroblasts [62]. FRNK overexpression abrogated the increased cell migration, and the increased FAK activation, in IPF lung fibroblasts [62]. In conclusion, data from patients showed that persistently activated adhesion and adhesive signaling, including FAK activation, is a hallmark of fibrotic cells.
3.5 FAK in Animal Models of Fibrosis
Knockout of the FAK gene in mice is lethal [58]. Studies performed with conditional cell-specific inactivation of the FAK gene in mice have provided useful insights into the role of FAK in fibrosis in vivo. In normal mouse skin, low levels of FAK phosphorylation are detected. However, it has been reported that FAK is activated after cutaneous injury and skin scarring [63]. Previous studies using keratinocyte-specific FAK knockout mice reported no wound healing phenotype [64]. Alternatively, fibroblast-specific FAK conditional knockout mice subjected to a hypertrophic scar-like mouse model of cutaneous scarring demonstrated less fibrogenesis when compared with wild-type mice [63]. The present authors and others have recently showed that FAK expression and activity are up-regulated in fibrotic foci in animal models of fibrosis, suggesting a role for FAK in vivo to promote lung fibrosis [37, 45]. Thus we have demonstrated that pharmacologic or genetic inactivation of FAK resulted in marked attenuation of lung fibrosis in a mouse model [45]. Similarly, in vivo administration of AG1879, a dual protein kinase inhibitor of PKB/Akt and FAK, could inhibit lung fibrogenesis in vivo [37]. Additionally, FRNK knockout mice displayed increased fibrogenesis in response to a pulmonary fibrotic stimulus in vivo, as compared with wild type mice, suggesting that FRNK is an endogenous inhibitor of FAK signaling in vivo [46].
4 Therapeutic Potential of FAK Inhibition in Fibrotic Disorders
Fibrotic diseases are largely initiated by chronic inflammatory processes. Thus, early studies focused on the effects of corticosteroids with or without immunosuppressive drugs, because of their known anti-inflammatory effects. Unfortunately, a large number of trials have shown little or no effect of these drugs on the progression of fibrosis. To date, it is widely speculated that the key effector cell in fibrogenesis is the myofibroblast. Given the pivotal role of FAK in fibroblast activation, drugs targeting FAK actions are thought to be beneficial in counteracting fibrosis.
A variety of in vitro and in vivo studies using murine models of fibrotic diseases suggest that FAK inhibitors exhibit potent antifibrotic effects (Tables 1, 2). In recent years, several orally bioavailable adenosine triphosphate (ATP)-competitive FAK inhibitors have been developed by pharmaceutical companies and have entered into early human clinical trials [65, 66]. One of the first clinically available specific FAK inhibitors was PF-562,271, which inhibited FAK phosphorylation in vivo in a dose-dependent fashion in several human subcutaneous xenograft models [67]. Recently, the present authors showed that PF-562,271 also prevented bleomycin-induced lung fibrosis in a mouse model [45]. The phase I study using PF-562,271 was performed in patients with head and neck, prostatic, and pancreatic cancer (NCT00666926, http://clinicaltrials.gov/). Clinically, PF-562,271 prolonged disease stabilization in a subgroup of patients. Due to the low toxicity of this drug, combination therapies with blocking antibodies or antagonists/inhibitors of profibrotic factor receptors seem possible. To note, PF-04554878 (NCT00787033, http://clinicaltrials.gov/) and GSK2256098 (NCT00996671 and NCT01138033, http://clinicaltrials.gov/) are being evaluated in phase I clinical trials in healthy volunteers and cancer patients. No final results have been reported so far.
The utilization of FAK inhibitors, initially developed as anticancer drugs, may have several rationales in fibrotic conditions, e.g., targeting of separate profibrotic factor signaling such as TGF-β1, ET-1, and cell microenvironment signals; or inhibiting different pathologic processes such as myofibroblast differentiation, fibroblast migration, and fibroblast resistance to anoikis. Thus, the possible indication for FAK inhibitors may lie in the prevention of the formation, invasion, and/or recruitment of collagen-producing cells, independent of their tissue/cell origin. However, to our knowledge, there are no clinical studies that have reported the effects of FAK inhibitors in any fibrotic diseases. Thus, FAK inhibitors should be considered for clinical trials within the next few years in order to elucidate whether inhibition of the FAK pathway is efficacious for the treatment of human fibrotic disorders. Our preclinical results suggest that PF-562,271, which has thus far been well tolerated in healthy volunteers and cancer patients, may offer therapeutic power for the treatment of fibrosis-related diseases.
5 Combination Regimens with Other Targeted Therapies
Since FAK interacts with other signaling molecules and pathways (Figs. 1, 2), there are potential promising combinatory treatment options of FAK inhibitors and other drugs. Recent in vitro studies identified novel non-canonical smad TGF-β1 targets including FAK that are activated by TGF-β1 in fibroblasts [36, 39, 46, 47, 53]. Interestingly, FAK regulates the formation of a tripartite membrane signaling complex in tumor cells that includes both TGF-β1 receptors and integrins [68]. Thus, combined FAK and TGF-β1 signaling inhibition may therefore have a biologic rationale. In fact, GC1008, which is a monoclonal antibody against TGF-β1 (NCT00356460, http://clinicaltrials.gov/), and PF-03446962, an antibody against one class I TGF-β receptor (NCT00557856, http://clinicaltrials.gov/), are in phase I testing. All together, these findings suggest that combined inhibition of TGF-β signaling and FAK, even in dose-sparing protocols, might be effective treatments in fibrogenesis. Reciprocal TGF-β-integrin signaling is normally implicated in a variety of pathologic processes including fibrogenesis [69, 70]. Accumulating evidence indicates that crosstalk between integrins and TGF-β signaling results in FAK activation [68]. Importantly, β1 integrin has been implicated in fibrosis and PF-04605412, a monoclonal antibody against α5β1 integrin, is being evaluated in phase I clinical trials in healthy volunteers and cancer patients (NCT009152783, http://clinicaltrials.gov/) [48, 70, 71]. Thus, combinatorial targeting of FAK and integrins may have a rationale to counteract human fibrogenesis.
Inhibition of intracellular pro-survival signal pathways may enhance the efficacy of FAK blockade. In this regard, due to its mutual activation, it is well known to have a synergistic effect of FAK and Src inhibition in cancer cells to promote cell apoptosis [72, 73]. Several small molecule inhibitors of Src including dasatinib are currently being investigated in clinical trials (NCT009152783, http://clinicaltrials.gov/). Recent data have described independent effects of the FAK and PKB/Akt pathway to trigger cell anoikis resistance [53]. Thus, combinatorial FAK and PKB/Akt treatments may have a rationale to induce myofibroblasts apoptosis. To note, Akt oral inhibitors are being evaluated in phase I clinical trials in healthy volunteers and cancer patients (NCT01266954, http://clinicaltrials.gov/).
6 Conclusion
Currently, there is no appropriate therapy available to counteract fibrotic disorders. The complexity of pathways operating to modulate fibroblast activation is only now becoming apparent. It is now known that the microenvironment plays an active role in fibroblast activation, hence in wound healing and fibrogenesis. Within this environment, fibroblasts respond to a host of signals including profibrotic growth factors such as TGFβ, ET-1, and CTGF and chemotactic factors such as LPA and PDGF, as well as signals from the extracellular matrix (Fig. 2). Targeting the pathways that mediate many of these signals has been a major goal in the effort to develop antifibrotic therapeutics. We emphasize that FAK is an attractive target for the therapy of fibrosis and needs further investigation.
Notes
Smad family member 3: the SMAD proteins are homologs of both the Drosophila protein, mothers against decapentaplegic (MAD) and the Caenorhabditis elegans protein SMA (from gene sma for small body size).
References
Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157(4 Pt 1):1301–15.
Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117(3):557–67.
Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117(3):524–9.
Gabbiani G. The myofibroblast: a key cell for wound healing and fibrocontractive diseases. Prog Clin Biol Res. 1981;54:183–94.
Chen SJ, et al. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. J Invest Dermatol. 1999;112(1):49–57.
Pannu J, et al. Increased levels of transforming growth factor beta receptor type I and up-regulation of matrix gene program: a model of scleroderma. Arthritis Rheum. 2006;54(9):3011–21.
Ishida W, et al. Intracellular TGF-beta receptor blockade abrogates Smad-dependent fibroblast activation in vitro and in vivo. J Invest Dermatol. 2006;126(8):1733–44.
Sime PJ, et al. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100(4):768–76.
Lagares D, et al. Endothelin 1 contributes to the effect of transforming growth factor beta1 on wound repair and skin fibrosis. Arthritis Rheum. 2010;62(3):878–89.
Serini G, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142(3):873–81.
Shi-Wen X, et al. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol. 2007;26(8):625–32.
Shi-Wen X, et al. Constitutive ALK5-independent c-Jun N-terminal kinase activation contributes to endothelin-1 overexpression in pulmonary fibrosis: evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Mol Cell Biol. 2006;26(14):5518–27.
Liu F, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190(4):693–706.
Mimura Y, et al. Constitutive phosphorylation of focal adhesion kinase is involved in the myofibroblast differentiation of scleroderma fibroblasts. J Invest Dermatol. 2005;124(5):886–92.
Kanner SB, et al. Monoclonal antibodies to individual tyrosine-phosphorylated protein substrates of oncogene-encoded tyrosine kinases. Proc Natl Acad Sci USA. 1990;87(9):3328–32.
Hanks SK, et al. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci USA. 1992;89(18):8487–91.
Kornberg L, et al. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J Biol Chem. 1992;267(33):23439–42.
Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68.
Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116(Pt 8):1409–16.
Girault JA, et al. The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends Biochem Sci. 1999;24(2):54–7.
Sieg DJ, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–56.
Streblow DN, et al. Human cytomegalovirus chemokine receptor US28-induced smooth muscle cell migration is mediated by focal adhesion kinase and Src. J Biol Chem. 2003;278(50):50456–65.
Schaller MD, et al. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J Cell Biol. 1995;130(5):1181–7.
Hildebrand JD, Schaller MD, Parsons JT. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp 125FAK, to cellular focal adhesions. J Cell Biol. 1993;123(4):993–1005.
Schaller MD, Borgman CA, Parsons JT. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp 125FAK. Mol Cell Biol. 1993;13(2):785–91.
Toutant M, et al. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol Cell Biol. 2002;22(22):7731–43.
Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15(2):954–63.
Schaller MD, et al. Autophosphorylation of the focal adhesion kinase, pp 125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680–8.
Owen JD, et al. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19(7):4806–18.
Owens LV, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55(13):2752–5.
Zhang C, et al. Focal adhesion kinase expressed by nerve cell lines shows increased tyrosine phosphorylation in response to Alzheimer’s A beta peptide. J Biol Chem. 1994;269(41):25247–50.
Shahrara S, et al. Differential expression of the FAK family kinases in rheumatoid arthritis and osteoarthritis synovial tissues. Arthritis Res Ther. 2007;9(5):R112.
Franchini KG, Clemente CF, Marin TM. Focal adhesion kinase signaling in cardiac hypertrophy and failure. Braz J Med Biol Res. 2009;42(1):44–52.
Rice DC, et al. Src autophosphorylation is an early event in pressure-mediated signaling pathways in isolated resistance arteries. Hypertension. 2002; 39(2 Pt 2): 502–7.
Morla AO, Mogford JE. Control of smooth muscle cell proliferation and phenotype by integrin signaling through focal adhesion kinase. Biochem Biophys Res Commun. 2000;272(1):298–302.
Thannickal VJ, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278(14):12384–9.
Vittal R, et al. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166(2):367–75.
Shi-wen X, et al. Requirement of transforming growth factor beta-activated kinase 1 for transforming growth factor beta-induced alpha-smooth muscle actin expression and extracellular matrix contraction in fibroblasts. Arthritis Rheum. 2009;60(1):234–41.
Liu S, et al. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell. 2007;18(6):2169–78.
Hayashida T, et al. MAP-kinase activity necessary for TGFbeta1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J Cell Sci. 2007;120(Pt 23):4230–40.
Dun ZN, et al. Specific shRNA targeting of FAK influenced collagen metabolism in rat hepatic stellate cells. World J Gastroenterol. 2010;16(32):4100–6.
Deng B, et al. Focal adhesion kinase mediates TGF-beta1-induced renal tubular epithelial-to-mesenchymal transition in vitro. Mol Cell Biochem. 2010;340(1–2):21–9.
Greenberg RS, et al. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 2006;20(7):1006–8.
Swigris JJ, Brown KK. The role of endothelin-1 in the pathogenesis of idiopathic pulmonary fibrosis. BioDrugs. 2010;24(1):49–54.
Lagares D, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64(5):1653–64.
Ding Q, et al. Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J Biol Chem. 2008;283(40):26839–49.
Thomas PE, et al. PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L417–28.
Liu S, et al. Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis Rheum. 2009;60(9):2817–21.
Kulkarni AA, et al. PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the PI3 K/Akt pathway: implications for therapy of fibrosis. PLoS One. 2011;6(1):e15909.
Jelaska A, Korn JH. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 2000;43(10):2230–9.
Xia H, et al. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. 2004;279(31):33024–34.
Wen LP, et al. Cleavage of focal adhesion kinase by caspases during apoptosis. J Biol Chem. 1997;272(41):26056–61.
Horowitz JC, et al. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19(4):761–71.
Reif S, et al. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem. 2003;278(10):8083–90.
Xia H, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. 2008;205(7):1659–72.
Horowitz JC, et al. Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. Int J Biochem Cell Biol. 2012;44(1):158–69.
Pardo A, Selman M. Molecular mechanisms of pulmonary fibrosis. Front Biosci. 2002;7:d1743–61.
Ilic D, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377(6549):539–44.
Kumagai N, et al. Lysophosphatidic acid induces tyrosine phosphorylation and activation of MAP-kinase and focal adhesion kinase in cultured Swiss 3T3 cells. FEBS Lett. 1993;329(3):273–6.
Chen HC, Guan JL. Stimulation of phosphatidylinositol 3’-kinase association with foca adhesion kinase by platelet-derived growth factor. J Biol Chem. 1994;269(49):31229–33.
Tangkijvanich P, et al. Platelet-derived growth factor-BB and lysophosphatidic acid distinctly regulate hepatic myofibroblast migration through focal adhesion kinase. Exp Cell Res. 2002;281(1):140–7.
Cai GQ, et al. Downregulation of FAK-related non-kinase mediates the migratory phenotype of human fibrotic lung fibroblasts. Exp Cell Res. 2010;316(9):1600–9.
Wong VW, et al. Focal adhesion kinase links mechanical force to skin fibrosis via inflammatory signaling. Nat Med. 2012;18(1):148–52.
Essayem S, et al. Hair cycle and wound healing in mice with a keratinocyte-restricted deletion of FAK. Oncogene. 2006;25(7):1081–9.
Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2010;19(6):777–88.
Schultze A, Fiedler W. Clinical importance and potential use of small molecule inhibitors of focal adhesion kinase. Anticancer Agents Med Chem. 2011;11(7):593–9.
Roberts WG, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68(6):1935–44.
Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11(5):R68.
Garamszegi N, et al. Extracellular matrix-induced transforming growth factor-beta receptor signaling dynamics. Oncogene. 2010;29(16):2368–80.
Kim KK, et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119(1):213–24.
Liu S, et al. Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo. J Cell Sci. 2010;123(Pt 21):3674–82.
Planas-Silva MD, et al. Role of c-Src and focal adhesion kinase in progression and metastasis of estrogen receptor-positive breast cancer. Biochem Biophys Res Commun. 2006;341(1):73–81.
Bolos V, et al. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco Targets Ther. 2010;3:83–97.
Hong S, et al. The role of focal adhesion kinase in the TGF-beta-induced myofibroblast transdifferentiation of human Tenon’s fibroblasts. Korean J Ophthalmol. 2012;26(1):45–8.
Dalla Costa AP, et al. FAK mediates the activation of cardiac fibroblasts induced by mechanical stress through regulation of the mTOR complex. Cardiovasc Res. 2010; 86(3): 421–31.
Chan MW, et al. FAK, PIP5KIgamma and gelsolin cooperatively mediate force-induced expression of alpha-smooth muscle actin. J Cell Sci. 2009;122(Pt 15):2769–81.
DiMichele LA, et al. Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ Res. 2006;99(6):636–45.
Peng X, et al. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116(1):217–27.
Clemente CF, et al. Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ Res. 2007;101(12):1339–48.
Acknowledgments
No sources of funding were used to conduct this study or prepare this manuscript.
Conflict of interest
The authors have no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lagares, D., Kapoor, M. Targeting Focal Adhesion Kinase in Fibrotic Diseases. BioDrugs 27, 15–23 (2013). https://doi.org/10.1007/s40259-012-0003-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-012-0003-4