
Abstract
Neutrophilic dermatoses constitute a heterogeneous group of dermatologic diseases, which are unified by the predominance of neutrophils within the inflammatory infiltrate on histopathology. The aims of this review were to provide an update on the clinical and histologic presentation of the main neutrophilic dermatoses and to develop a guide for clinical practice. A structured literature search of PubMed, Medline, and Embase was performed, using the key words “neutrophilic disorders”, “cutaneous small vessel vasculitis”, “Sweet’s syndrome”, “bowel associated dermatosis arthritis syndrome”, “Behcet’s”, “palisaded neutrophilic and granulomatous dermatosis”, “rheumatoid neutrophilic dermatitis”, and “pyoderma gangrenosum”. Related articles were screened for key terms and were included if appropriate. This group contains a wide spectrum of unique disorders, each with its own histologic and clinical subtleties, making specific diagnosis of a given entity within the group diagnostically challenging. The fact that overlapping forms of neutrophilic dermatoses, which share features of multiple neutrophilic dermatoses, are not uncommon makes the diagnoses more challenging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Neutrophilic dermatoses are a heterogeneous group of disorders characterized by infiltration of neutrophils in active lesions. |
The risk of association with underlying conditions attracts attention toward early diagnosis of neutrophilic dermatoses. |
1 Introduction
Neutrophilic dermatoses constitute a heterogeneous group of noninfectious dermatologic diseases, which are unified by the predominance of neutrophils within the inflammatory infiltrate of active skin lesions. While a publication by Jorizzo et al. in 1988 was the first to unify and describe a classification of neutrophilic reactions as distinct but related entities, subsequent research has shown that these entities have significant overlap in histopathologic and clinical findings, and can occur concurrently in the same patient [1]. Indeed, overlapping forms of neutrophilic dermatoses, which share features of multiple neutrophilic dermatoses, are not infrequently encountered [2]. A structured literature search of PubMed, Medline, and Embase from 1988 to the present was performed, using the key words “neutrophilic disorders”, “cutaneous small vessel vasculitis”, “Sweet’s syndrome”, “bowel associated dermatosis arthritis syndrome”, “Behcet’s”, “palisaded neutrophilic and granulomatous dermatosis”, “rheumatoid neutrophilic dermatitis”, and “pyoderma gangrenosum”. The abstracts and relevant full-text articles were reviewed. This review provides an update on the clinical and histologic presentations of the main prototypic neutrophilic dermatoses (Table 1).
2 Cutaneous Small-Vessel Vasculitis
2.1 Clinical Presentation
Cutaneous small-vessel vasculitis (CSVV) is classified as a subset of vasculitis that predominantly affects small vessels, including intraparenchymal arteries, arterioles, capillaries, and venules [3, 4]. While CSVV is frequently associated with a wide spectrum of systemic inflammatory conditions, malignancies, infections, or drug hypersensitivities [5], approximately half of all cases are idiopathic.
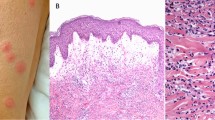
Clinically, CSVV presents as purpura, often palpable purpuric papules, or non-blanching macules, and favors dependent areas, areas of trauma, and areas of pressure, such as under tight-fitting clothing (Fig. 1a) [6–8].

Leukocytoclastic vasculitis: a palpable purpura, which may evolve to ulceration as superficial infarctions occur; b histology shows neutrophilic infiltration around and within dermal blood vessels and fibrinoid necrosis of blood vessel walls
In general, the presence of constitutional signs or symptoms—such as joint pain or swelling; abdominal pain, melena, frothy or discolored urine, or abnormal urinalysis; focal peripheral sensory or motor nerve problems or central nerve deficits—should all prompt consideration for systemic involvement in vasculitis. The presence of paresthesia or the absence of painful lesions and hypocomplementemia have been identified as risk factors for an associated systemic disease [6, 7, 9]. Given the above associations, all patients with CSVV must be evaluated for internal involvement, especially of the kidneys, gastrointestinal tract, and joints, which requires a thorough history, a detailed and targeted physical examination, and a thorough laboratory workup, which often includes urinalysis, a complete blood count with differential, creatinine level, complement 3 and 4 levels, and fecal occult blood.
2.2 Histopathology
The hallmark histopathologic pattern of CSVV is leukocytoclastic vasculitis, which is characterized by angiocentric segmental inflammation, endothelial cell swelling, and fibrinoid necrosis of blood vessel walls (postcapillary venules). The cellular infiltrate around and within dermal blood vessel walls is composed mainly of neutrophils, many of which exhibit fragmentation of nuclei (karyorrhexis or leukocytoclasia) [Fig. 1b]. Palpable purpura can be explained by infiltration of leukocytes (palpability) and the resulting extravasation of red blood cells from the damaged blood vessel (purpura).
Direct immunofluorescence is positive in around 80 % of cases, demonstrating deposition of complement C3, immunoglobulin (Ig)-M, IgA, and/or IgG (generally in that order of frequency) in a granular pattern within the vessel walls [10].
2.3 Pathogenesis
The pathogenesis of CSVV has not been fully elucidated, but it includes circulating immune complex deposition, increased adhesiveness between inflammatory cells and the endothelium, and neutrophil-mediated damage to postcapillary venules. Although 50 % of cases are idiopathic and follow a benign, self-limited course, an attempt to address underlying systemic involvement or causes should be made. This is best done through a multidisciplinary, collaborative approach.
2.4 What’s New?
In recent decades, additional etiologies have been implicated and associated with CSVV, including drugs [11, 12], infections, malignancies [13], herbal supplements [14], and systemic diseases. It is worth remembering that association does not prove causation. Therefore, each patient’s history needs to be assessed carefully.
The Chapel Hill Consensus Conference statement replaced the name “Henoch–Schönlein purpura” with “IgA vasculitis” because of evidence that IgA deposits in vessel walls are the defining feature [4]. Also, the new category of single-organ vasculitis can occur with IgA vasculitis. Recent studies have suggested that IgA vasculitis in adults may represent a paraneoplastic vasculitis, with renal cancer being the top culprit [15].
Notably, a lymphocytic form of small-vessel vasculitis has also been described in cases of collagen vascular disease [14, 16, 17]. In a study by Bahrami et al. [18] in 60 patients with CSVV, marked tissue eosinophilia was suggested as a reliable indicator of drug-induced cases [19, 20].
3 Sweet’s Syndrome
3.1 Clinical Presentation
Sweet’s syndrome, or acute febrile neutrophilic dermatosis, is the prototypic member of the neutrophilic dermatoses. While up to 50 % of cases are idiopathic, up to 35 % of patients have underlying internal malignancies [21]. These are predominantly hematologic malignancies, particularly myelodysplastic syndrome and acute myelogenous leukemia, although 7–15 % of these patients have solid malignancies, the most common of which are genitourinary tumors [22, 23]. Other associations include postinfectious, inflammatory autoimmune disorders, as well as reactions to an ever increasing list of medications.
Clinically, Sweet’s syndrome typically develops several days after an upper respiratory tract infection or occasionally after acute viral gastroenteritis. It typically presents as bilateral, edematous, tender plaques or nodules in the setting of fever and arthralgia. Interestingly, a recent study of 77 patients found that only 39 and 27 % of patients reported a fever and arthralgia, respectively [21]. The lesions favor the head, neck, and upper and lower extremities, and occasionally display a central yellowish discoloration, which may create a targetoid appearance (Fig. 2a). Cohen [24] has noted that multiple neutrophilic dermatoses may exhibit an annular pattern. Pathergy may be observed, with the appearance of new lesions at the site of trauma. Sweet’s syndrome lesions can uncommonly involve internal organs, especially in the setting of an underlying malignancy. The most commonly affected areas include the eyes, neuromuscular system, joints, kidneys, lungs, heart, and liver [23, 24]. Patients with the presence of (a) vesiculobullous lesions, particularly ones that progress to pyoderma gangrenosum (PG)-like ulcers; (b) a histiocytoid clinicopathologic variant; or (c) extensive, asymmetrical distribution of lesions appear to have a higher propensity for an underlying malignancy. The criteria for the diagnosis of Sweet’s syndrome are listed in Tables 2 and 3 [25, 26].

Febrile neutrophilic dermatosis: a tender erythematous plaques on the hands; b characteristic histopathology findings include significant edema with diffuse nodular and perivascular neutrophilic infiltration
3.2 Histopathology
The characteristic histolopathologic finding of a classic lesion is a diffuse pan-dermal or nodular and perivascular dermal neutrophilic infiltrate, although infiltration of the appendages may also be seen. Superficial dermal edema is usually seen (Fig. 2b). While most cases of Sweet’s syndrome present with leukocytoclasia, dilation of small blood vessels, fragmentation of neutrophil nuclei without fibrin deposition, and neutrophils within vessel walls [27], true vasculitis has been seen in several cases [10, 11], and its presence should not rule out the diagnosis of Sweet’s syndrome.
3.3 Pathogenesis
While the exact etiology of Sweet’s syndrome remains unknown, circulating autoantibodies, cytokines, dermal dendrocytes, immune complexes, and genetic predisposition [28] have been discussed in the pathogenesis of Sweet’s syndrome [29] and suggest an underlying hypersensitivity reaction to a variety of stimuli. Indeed, numerous associations capable of inducing these responses, including hematologic malignancies (lymphoma, myeloma) and solid organ malignancies, drugs, and inflammatory diseases (inflammatory bowel disease, autoimmune connective tissue disorders, Behcet’s disease, thyroid disease, sarcoidosis) have been described [30].
3.4 What’s New?
Sweet’s syndrome has been expanded in terms of the spectrum of its etiologic, clinical, and histologic manifestations. There are now numerous recognized skin variants of Sweet’s syndrome, and several internal organs have been shown to develop lesions consistent with Sweet’s syndrome. A variant of Sweet’s syndrome, termed “histiocytoid Sweet’s syndrome” [31, 32], has recently been described, though that entity may be better characterized as a form of leukemia cutis with immature cells [33]. Currently, Sweet’s syndrome is classified as either idiopathic or secondary to drugs, autoimmune conditions, or malignancies. Diagnostic criteria have been formulated for classic as well as drug-induced Sweet’s syndrome (Tables 2, 3) [25, 26]. In addition, several new treatment modalities have been described, which include potassium iodide, dapsone, colchicine, indomethacin, clofazimine, cyclosporine, interferon-α, and tumor necrosis factor (TNF)-blocking biologics [34].
Overall, Sweet’s syndrome typically follows a benign course, which, if untreated, can involute spontaneously without scarring in 5–12 weeks. However, there is a significant proportion of patients in whom the disease affects vital internal organs or is associated with an increasing number of malignancies or drugs. Given the febrile nature of Sweet’s syndrome, patients are often uncomfortable, and treatment may be necessary for symptomatic relief. Patients with malignancy-associated Sweet’s syndrome, particularly patients with myelodysplastic syndrome/acute myelogenous leukemia, may be mistakenly diagnosed with infection. In these cases, diagnosis and treatment of Sweet’s syndrome may be important to resolve the fever. Sweet’s syndrome is rapidly and strikingly responsive to systemic corticosteroids (as one of the minor criteria), and treated patients will often defervesce within 24 h. Patients who fail to respond warrant consideration for alternate diagnoses. In all patients with Sweet’s syndrome, it is important to perform a careful review of systems, along with a targeted investigation.
4 Bowel-Associated Dermatosis–Arthritis Syndrome
4.1 Clinical Presentation
Bowel-associated dermatosis–arthritis syndrome (BADAS), previously known as bowel bypass syndrome, is usually seen subsequent to gastrointestinal pathology [35]. The first cases were reported in 1971 following jejunoileal bypass surgery. Dicken and Seehafer in 1979 coined the term “bowel bypass syndrome” and related the pathogenesis to the bacterial overgrowth in blind loops of bowel [36, 37]. Subsequently, following the reporting of cases of this syndrome in patients who had no previous history of intestinal bypass surgery but had inflammatory bowel disease or Bilroth II surgery with blind loops of bowel, Jorizzo introduced the term “bowel-associated dermatosis–arthritis syndrome” [38].
BADAS typically presents with polyarthralgia, polyarthritis, myalgia, and a variety of cutaneous manifestations, in conjunction with significant bowel symptoms, including diarrhea and malabsorption. Skin manifestations include panniculitis, ecchymosis, pustular vasculitis, and erythema nodosum (EN). Skin lesions often present rapidly within days and may recur over several weeks. The polyarthritis is episodic and migratory, and involves predominantly peripheral joints [39].
4.2 Histopathology
Early lesions demonstrate infiltration of neutrophils in mid-dermal venules and a perivascular mononuclear cell infiltrate. In older lesions, the edema in the papillary dermis increases to form a vesicle and the density of the dermal neutrophil infiltrate increases to form a pustule. There is no endothelial necrosis, infarction, or fibrinoid degeneration characteristic of a true leukocytoclastic vasculitis [39].
4.3 Pathogenesis
The pathogenesis of BADAS is poorly understood, but it is believed that blind loops of bowel and microbial overgrowth lead to an immune complex disease due to bacterial antigens. The disease is purported to be associated with entities that predispose to bacterial overgrowth secondary to bowel insults that promote stasis. These include surgery (gastric resection, jejunoileal bypass, biliopancreatic diversion, blind loops of bowel) or inflammatory conditions (inflammatory bowel disease, diverticulitis, peptic ulcer disease), and there have even been reports after appendicitis [40]. BADAS has been described between 3 months to 5 years post-surgery and with a reported incidence of up to 20 %. Typically, flu-like symptoms precede the development of the skin rash and arthritis by 12–36 h [39].
4.4 What’s New?
In recent decades, multiple cases of BADAS have been reported, and the characteristics and etiology have been expanded and better defined. With the increase in the incidence of obesity and bariatric surgeries related to this, case reports of BADAS following this type of surgery have been reported rarely [37, 40]. The delayed period of time between surgery and development of BADAS can hinder prompt diagnosis and treatment of the disease.
5 Behcet’s Disease
5.1 Clinical Presentation
Behcet’s disease is an inflammatory multisystem disease, which can have an extremely variable course with unpredictable exacerbations and remissions. The exact pathogenic mechanism is unknown, but it is believed to involve genetic and environmental factors, with the HLA-B*51 allele being strongly associated with the disease in Asian populations [41]. Behcet’s disease is typified by the presence of recurrent oral and genital ulceration, ocular abnormalities (uveitis, retinal vasculitis), and skin lesions [42]. Cutaneous findings have been reported in 80 % of patients, commonly on the face and acral sites [2]. Recurrent oral and genital ulcers, pseudo-folliculitis, pustular and purpuric papules, PG, and EN-like lesions may be seen. EN-like lesions may be seen on the buttocks and legs, particularly in women, as well as on the face and neck, and can resemble thrombophlebitis—which itself can affect 30 % of patients with Behcet’s disease. The various systems are affected by both vasculitis and thrombotic events. A pathergy test is positive in 30–40 % of patients with Behcet’s disease and more commonly in patients of Asian or Middle Eastern ethnicity [29, 43, 44].
5.2 Histopathology
Behcet’s disease is a systemic perivasculitis with a variety of histologic findings. In early lesions, significant neutrophil infiltration is seen, including in mucocutaneous aphthae, the skin pathergy reaction, nodular lesions of the skin, and ocular lesions [41].
5.3 Pathogenesis
The exact pathophysiologic mechanism is still undetermined; however, studies have demonstrated the potential role of impaired fibrinolytic kinetics in generation of the hypercoagulable/prothrombotic state in Behcet’s disease [41]. Molecular mimicry following infection with various pathogens and immunologic abnormality likely play a significant role [29]. Multiple cytokines and chemokines play a substantial role in the disease pathogenesis and even in organ lesions, which may provide essential clues to the target therapy regimen of cytokine agents [45]. Interleukin (IL)-6 has been shown to be associated with disease activity in several studies [45].
5.4 What’s New?
In the revised Chapel Hill Consensus Conference classification, Behcet’s disease has been classified as variable vessel vasculitis (VVV), with multiple presentations such as small-vessel vasculitis, arteritis, arterial aneurysms, and venous and arterial thromboangiitis and thrombosis [4]. Recent studies have demonstrated the role of cytokines in the pathogenesis of Behcet’s disease. The cytokines involved can be categorized as T helper (Th)-1, Th2, and Th17 types, chemokines, and other proinflammatory cytokines [45]. The IL-10 and IL-23/IL-17 pathways are crucial in the pathogenesis of the disease, and new treatments have been used with success, such as anti–IL-1 targeted inhibitors [46], anti–IL-6 receptor inhibitors [47], and anti-CD20 monoclonal antibodies, intravenous immunoglobulin, and stem cell treatment [48].
6 Palisaded Neutrophilic and Granulomatous Dermatosis
6.1 Clinical Presentation
Palisaded neutrophilic and granulomatous dermatosis (PNGD) is a rare neutrophilic dermatosis, with varied clinical presentations. It usually presents in patients with an underlying systemic condition, such as a connective tissue disease, inflammatory arthritis, lymphoproliferative disorder, or infection; in rare cases, it may be medication induced [49].
The most common clinical presentation is that of erythematous papules, which may appear to be umbilicated or crusted, and symmetrically distributed on extensor surfaces of the upper extremities, especially the fingers and elbows [50–54]. These have been reported as ranging from urticarial papules to more fixed pink-red papules to more flesh-colored fibrotic papules. In a review of 81 patients [51], the extremities were the most common presenting location, with 51.1 % involving the upper extremities and 27.7 % involving the lower extremities. The trunk and head and neck regions were involved in 21.2 % of patients [55–57].
While erythematous papules around the elbows are the classic description, the breadth of lesions now described as PNGD is quite varied and can also include patches, plaques, or nodules.
6.2 Histopathology
Like its clinical presentation, the histopathologic features of PNGD vary. Chu et al. [58] have suggested that it represents a histologic continuum. Early lesions show evidence of leukocytoclastic vasculitis, with diffuse pan-dermal infiltrates composed of neutrophils, nuclear debris, and strands of amorphous basophilic material. Late lesions usually lack leukocytoclasia [16], with palisaded granulomas and degenerated collagen being the predominant findings [58]. Each stage of development can elicit a different differential diagnosis; thus, clincopathologic correlation is compulsory.
6.3 Pathogenesis
The responsible underlying mechanisms for development of PNGD remain poorly understood. Direct immunofluorescence studies have demonstrated the presence of immunoglobulins and C3 deposits in vessel walls [50, 58]. PNGD may be a cutaneous marker of systemic disease, especially diseases with a suspected autoimmune pathogenesis [54]. PNGD can occur in the setting of rheumatoid arthritis [51, 54]. The presence of PNGD, like rheumatoid nodules and rheumatoid vasculitis, has been associated with higher disease activity scores and greater disease severity [59].
6.4 What’s New?
Different terms were used to describe this condition until the description “palisaded neutrophilic and granulomatous dermatitis of immune-complex disease” was coined in 1994. Systemic lupus erythematous and rheumatoid arthritis have been more commonly reported as underlying causes [54, 60]. PNGD and other reactive granulomatous skin findings have been reported in the setting of malignancies, particularly hematologic malignancies (myelodysplastic syndrome, leukemia, lymphoma, and paraproteinemia). If the underlying disease is not known, then the history, physical examination, and laboratory tests should be directed toward diagnosing the underlying conditions. There is confusion in the literature between PNGD and a related reactive granulomatous process, interstitial granulomatous dermatitis (IGD). Some authors consider PNGD to be a subtype of IGD, whereas others consider PNGD and IGD to be distinct reactive processes, which may be similar to or distinct from rheumatoid neutrophilic dermatitis (RND).
7 Rheumatoid Neutrophilic Dermatitis
7.1 Clinical Presentation
RND is an uncommon skin condition, which was first described by Ackerman in 1978 [59, 61, 62]. While most described cases are associated with RA, RND can also be observed in patients with seronegative arthritis [62, 65].
Patients classically present with papulonodules and/or plaques distributed on the extensor surfaces of the extremities (Fig. 3), particularly the hands and forearms, as well as the neck and trunk. Overall, the lesions tend to be asymptomatic, and patients do not display constitutional symptoms [63].

Rheumatoid neutrophilic dermatitis, presenting as papulonodules on the extensor surface of the hand
7.2 Histopathology
Histologically, one observes a dense neutrophilic dermal infiltrate, which is accentuated in the upper and middle levels. Indeed, in late lesions, this last finding may be the only evidence of a neutrophilic dermatosis; thus, this may make the diagnosis more difficult [63, 64]. Overall, histologic features of both Sweet’s syndrome and dermatitis herpetiformis, along with plasma cells, all in the context of rheumatoid arthritis, should lead to the diagnosis.
7.3 Pathogenesis
The pathogenesis and etiology of RND is not completely understood, though it has been suggested that it is likely an immune-complex–mediated disease [65].
7.4 What’s New?
Numerous skin variants of RND have been described. Overall, RND typically follows a benign course, and while the lesions may resolve spontaneously, the norm for this condition is to be persistent, symmetrical, and chronic. Some authors also view PNGD as yet another histologic variant of RND [54], though in the absence of visible granulomas on histology, it seems reasonable to suggest that the two represent separate entities.
8 Pyoderma Gangrenosum
8.1 Clinical Presentation
PG is a rare, recurring, chronic, and painful disease, with four distinct clinical variants: ulcerative, bullous, pustular, and superficial granulomatous (or vegetative). Although the cause of PG is unknown, between 45 and 75 % of cases are associated with systemic disease, including inflammatory bowel disease, myeloproliferative disorders, connective tissue disease, and rheumatoid arthritis [25, 43, 66, 67].
The clinical course of PG is unpredictable—from a precipitous onset with rapid spread to a more indolent pattern. PG lesions comprise one or more central papulopustules surrounded by an erythematous or violaceous gunmetal induration, a bulla on a violaceous base, or erythematous nodules (Fig. 4a). Pathergy is present in 25–30 % of patients with PG and presents as extension of existing PG lesions or appearance of new lesions following dermal injury [27, 43]. The vesiculobullous variant of PG has been associated with myeloproliferative disorders in as many as 70 % of cases and has an increased incidence of occurrence on the face and the dorsum of the hands; this may show significant overlap with Sweet’s syndrome and the subtype neutrophilic dermatosis of the dorsal hand [43, 68]. The pustular form is commonly associated with inflammatory bowel disease [69]. The vegetative form of PG is not usually associated with the underlying disease and is found predominantly on the head and neck [25, 43]. Very rarely, as in Sweet’s syndrome, extracutaneous sterile neutrophilic infiltrates can be observed in the bones, liver, lungs, pancreas, spleen, kidneys, and central nervous system of patients with PG. PG is often a diagnosis of exclusion, and frequently it is misdiagnosed [67]. The proposed diagnostic criteria for classic ulcerative PG are listed in Table 4 [70].

Pyoderma gangrenosum (PG): a painful ulcer with a purulent base and a gunmetal border; b neutrophilic infiltration of the dermis, which is characteristic of PG on histology
8.2 Histopathology
The hallmark of PG is accumulation of neutrophils in the skin and, rarely, in internal organs (Fig. 4b). Although PG is characterized by the presence of inflammatory dermal infiltrates composed of mature neutrophils on histology, the diagnosis is based on clinical correlation and exclusion of other causes.
8.3 Pathogenesis
Immunohistochemical studies have demonstrated the expression of CD3-positive T cells and CD163-positive cells (macrophages) at the wound edge, while, in contrast, the wound bed has mainly contained neutrophil markers. These findings demonstrate that activated T cells and macrophages at the wound edge may pave the way for extension of the ulcer [25].
8.4 What’s New?
The role of inflammasomes in the pathogenesis of some cases of PG has been highlighted in recent literature. There have been reports of PG being treated with biologics. New medications including anti-TNFs, such as infliximab (chimeric anti-TNF-α monoclonal antibody), adalimumab (humanized anti-TNF-α monoclonal antibody), and etanercept (fusion protein human p75 TNF-α receptor IgG1), have been used as treatment options, with some success [67].
Brooklyn et al. [71] demonstrated that infliximab at a dose of 5 mg/kg was superior to placebo for treatment of PG in a randomized, placebo controlled trial, where 29 patients received infliximab. At week 6, 20 patients (69 %) demonstrated a beneficial clinical response, with remission in 6 patients (21 %), while the control group had no response. PG has been listed in some literature as an auto-inflammatory disorder, especially in the context of PAPA (pyogenic arthritis, PG, and cystic acne) syndrome [2, 24]. In PAPA syndrome, different mutations induce assembly of inflammasomes through increased binding affinity to pyrin. Overproduction of IL-1β [27] triggers release of several proinflammatory cytokines and chemokines, which recruit and activate neutrophils and lead to neutrophil-mediated inflammation [25, 72].
Hadi and Lebwohl [73] showed that clinical clues to PG are undervalued. In the absence of clinical characteristics, pathologic features should not be the sole grounds for diagnosing an ulcer appearing at an atypical site. Misdiagnosis of PG is common. In a study by Weenig et al. [74], a retrospective chart review of 240 patients demonstrated a large number of cases of skin ulcers with resemblance to PG. These findings highlight the need for thorough evaluation in all patients with a diagnosis of PG in order to rule out alternative diagnoses.
9 Conclusion
Neutrophilic dermatoses are a heterogeneous group of reactive diseases, which are unified by the predominance of neutrophils within the inflammatory infiltrate of active skin lesions. A comprehensive review of the treatment options is beyond the scope of this article. The first intervention consists of supportive care and avoidance of possible triggers, which may be the only intervention needed by some patients. Additional treatments should be prescribed according to the severity of the cutaneous involvement, chronicity, and whether or not systemic involvement is present. Careful management of chronic underlying diseases—for example, rheumatoid arthritis or inflammatory bowel disease—can prevent rebound flares [3]. Despite the clinical overlap, each entity shows unique differences in the frequency and/or the hierarchy of associated systemic findings. These associations highlight the need for more attention toward this group of inflammatory diseases.
References
Jorizzo JL, Solomon AR, Zanolli MD, Leshin B. Neutrophilic vascular reactions. J Am Acad Dermatol. 1988;19(6):983–1005.
Berk DR, Bayliss SJ. Neutrophilic dermatoses in children. Pediatr Dermatol. 2008;25(5):509–19.
Kinney MA, Jorizzo JL. Small-vessel vasculitis. Dermatol Ther. 2012;25(2):148–57.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1–11.
Russell JP, Gibson LE. Primary cutaneous small vessel vasculitis: approach to diagnosis and treatment. Int J Dermatol. 2006;45(1):3–13.
Sais G, Vidaller A, Jucgla A, Servitje O, Condom E, Peyri J. Prognostic factors in leukocytoclastic vasculitis: a clinicopathologic study of 160 patients. Arch Dermatol. 1998;134(3):309–15.
Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis: clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol. 1984;120(4):484–9.
Caucanas M, Heylen A, Rolland F, Muller G, Olemans C, Sass U, et al. Associated pyoderma gangrenosum, erythema elevatum diutinum, and Sweet’s syndrome: the concept of neutrophilic disease. Int J Dermatol. 2013;52(10):1185–8.
Swerlick RA, Lawley TJ. Cutaneous vasculitis: its relationship to systemic disease. Med Clin N Am. 1989;73(5):1221–35.
Carlson JA, Ng BT, Chen KR. Cutaneous vasculitis update: diagnostic criteria, classification, epidemiology, etiology, pathogenesis, evaluation and prognosis. Am J Dermatopathol. 2005;27(6):504–28.
Peuvrel L, Quereux G, Brocard A, Saint-Jean M, Freour E, Josselin N, et al. Atypical cutaneous vasculitis under lapatinib. Eur J Dermatol. 2013;23(4):540–1.
Novoa RA, Honda K, Koon HB, Gerstenblith MR. Vasculitis and panniculitis associated with vemurafenib. J Am Acad Dermatol. 2012;67(6):e271–2.
Podjasek JO, Wetter DA, Pittelkow MR, Wada DA. Cutaneous small-vessel vasculitis associated with solid organ malignancies: the Mayo Clinic experience, 1996–2009. J Am Acad Dermatol. 2012;66(2):e55–65.
Ingraffea A, Donohue K, Wilkel C, Falanga V. Cutaneous vasculitis in two patients taking an herbal supplement containing black cohosh. J Am Acad Dermatol. 2007;56(5 Suppl):S124–6.
Podjasek JO, Wetter DA, Pittelkow MR, Wada DA. Henoch-Schonlein purpura associated with solid-organ malignancies: three case reports and a literature review. Acta Derm Venereol. 2012;92(4):388–92.
Lever WF, Schaumburg-Lever S. Histopathology of the skin. Philadelphia: JB Lippincott; 1990.
Zax RH, Hodge SJ, Callen JP. Cutaneous leukocytoclastic vasculitis: serial histopathologic evaluation demonstrates the dynamic nature of the infiltrate. Arch Dermatol. 1990;126(1):69–72.
Bahrami S, Malone JC, Webb KG, Callen JP. Tissue eosinophilia as an indicator of drug-induced cutaneous small-vessel vasculitis. Arch Dermatol. 2006;142(2):155–61.
Haeberle MT, Adams WB, Callen JP. Treatment of severe cutaneous small-vessel vasculitis with mycophenolate mofetil. Arch Dermatol. 2012;148(8):887–8.
Sokumbi O, Wetter DA, Makol A, Warrington KJ. Vasculitis associated with tumor necrosis factor-alpha inhibitors. Mayo Clin Proc. 2012;87(8):739–45.
Rochet NM, Chavan RN, Cappel MA, Wada DA, Gibson LE. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69(4):557–64.
Anzalone CL, Cohen PR. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Curr Opin Hematol. 2013;20(1):26–35.
Paydas S. Sweet’s syndrome: a revisit for hematologists and oncologists. Crit Rev Oncol/Hematol. 2013;86(1):85–95.
Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10(5):301–12.
Marzano AV, Ishak RS, Saibeni S, Crosti C, Meroni PL, Cugno M. Autoinflammatory skin disorders in inflammatory bowel diseases, pyoderma gangrenosum and Sweet’s syndrome: a comprehensive review and disease classification criteria. Clin Rev Allergy Immunol. 2013;45(2):202–10.
Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 Pt 2):918–23.
Bonamigo RR, Razera F, Olm GS. Neutrophilic dermatoses: part I. An Bras Dermatol. 2011;86(1):11–25 (quiz 26–7).
Parsapour K, Reep MD, Gohar K, Shah V, Church A, Shwayder TA. Familial Sweet’s syndrome in 2 brothers, both seen in the first 2 weeks of life. J Am Acad Dermatol. 2003;49(1):132–8.
Razera F, Olm GS, Bonamigo RR. Neutrophilic dermatoses: part II. An Bras Dermatol. 2011;86(2):195–209 (quiz 210–1).
Vanourny J, Swick BL. Sweet syndrome with systemic inflammatory response syndrome. Arch Dermatol. 2012;148:969–70.
Heymann WR. Histiocytoid Sweet syndrome. J Am Acad Dermatol. 2009;61(4):693–4.
Requena L, Kutzner H, Palmedo G, Pascual M, Fernandez-Herrera J, Fraga J, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141(7):834–42.
Chavan RN, Cappel MA, Ketterling RP, Wada DA, Rochet NM, Knudson R, et al. Histiocytoid Sweet syndrome may indicate leukemia cutis: a novel application of fluorescence in situ hybridization. J Am Acad Dermatol. 2014;70(6):1021–7.
Raza S, Kirkland RS, Patel AA, Shortridge JR, Freter C. Insight into Sweet’s syndrome and associated-malignancy: a review of the current literature. Int J Oncol. 2013;42(5):1516–22.
Halawai A, Abiad F, Abbas O. Bariatric surgery and its effects on the skin and skin diseases. Obes Surg. 2013;23:408–13.
Dicken CH, Seehafer JR. Bowel bypass syndrome. Arch Dermatol. 1979;115:837–9.
Tu J, Chan JJ, Yu LL. Bowel bypass syndrome/bowel-associated dermatosis arthritis syndrome post laparoscopic gastric bypass surgery. Aust J Dermatol. 2011;52:e5–7.
Jorizzo JL, Apisarnthanarax P, Subrt P, Hebert AA, Henry JC, Raimer SS, et al. Bowel-bypass syndrome without bowel bypass. Bowel-associated dermatosis-arthritis syndrome. Arch Int Med. 1983;143:457–61.
Slater GH, Kerlin P, Georghiou PR, Fielding GA. Bowel-associated dermatosis-arthritis syndrome after biliopancreatic diversion. Obes Surg. 2004;14:133–5.
Prpic-Massari L, Kastelan M, Brajac I, Cabrijan L, Zamolo G, Massari D. Bowel-associated dermatosis–arthritis syndrome in a patient with appendicitis. Med Sci Monit. 2007;13(8):CS97–100.
Mendoza-Pinto C, Garcia-Carrasco M, Jimenez-Hernandez M, Jimenez Hernandez C, Riebeling-Navarro C, Nava Zavala A, et al. Etiopathogenesis of Behcet’s disease. Autoimmun Rev. 2010;9:241–5.
Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M, et al. Behcet’s disease: from East to West. Clin Rheumatol. 2010;29:823–33.
Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M, et al. Behcet’s disease in Iran: analysis of 6500 cases. Int J Rheum Dis. 2010;13(4):367–73.
Hatemi G, Seyahi E, Fresko I, Hamuryudan V. Behcet’s syndrome: a critical digest of the recent literature. Clin Exp Rheumatol. 2012;30(3 Suppl 72):S80–9.
Zhou ZY, Chen SL, Shen N, Lu Y. Cytokines and Behcet’s disease. Autoimmun Rev. 2012;11:699–704.
Emmi G, Silvestri E, Ciucciarelli L, Squatrito D, Emmi L. Anti-IL1 blocking agents in drug-resistant Behcet’s syndrome: our little case series. Clin Exp Rheum. Epub 2014 Apr 7.
Urbaniak P, Hasler P, Kretzschmar S. Refractory neuro-Behcet treated by tocilizumab: a case report. Clin Exp Rheumatol. 2012;30(3 Suppl 72):S73–5.
Ozen S, Eroglu FK. Pediatric-onset Behcet disease. Curr Opin Rheumatol. 2013;25(5):636–42.
Wilmoth GJ, Perniciaro C. Cutaneous extravascular necrotizing granuloma (Winkelmann granuloma): confirmation of the association with systemic disease. J Am Acad Dermatol. 1996;34(5 Pt 1):753–9.
Kim SK, Park CK, Park YW, Jun JB, Yoo DH, Bae SC. Palisaded neutrophilic granulomatous dermatitis presenting as an unusual skin manifestation in a patient with Behcet’s disease. Scand J Rheumatol. 2005;34(4):324–7.
Hantash BM, Chiang D, Kohler S, Fiorentino D. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58(4):661–4.
Kreuter A, Rose C, Zillikens D, Altmeyer P. Bullous rheumatoid neutrophilic dermatosis. J Am Acad Dermatol. 2005;52(5):916–8.
Fujio Y, Funakoshi T, Nakayama K, Amagai M, Ohyama M. Rheumatoid neutrophilic dermatosis with tense blister formation: a case report and review of the literature. Australas J Dermatol. 2012;.;55((1):):e12–4.
Sangueza OP, Caudell MD, Mengesha YM, Davis LS, Barnes CJ, Griffin JE, et al. Palisaded neutrophilic granulomatous dermatitis in rheumatoid arthritis. J Am Acad Dermatol. 2002;47(2):251–7.
Biswas A, Chittari K, van Gey Pittius D, Stephens M, Tan BB. Palisaded neutrophilic and granulomatous dermatitis in a child with type I diabetes mellitus and coeliac disease. Br J Dermatol. 2008;159(2):488–9.
Germanas JP, Mehrabi D, Carder KR. Palisaded neutrophilic granulomatous dermatitis in a 12-year-old girl with systemic lupus erythematosus. J Am Acad Dermatol. 2006;55(2 Suppl):S60–2.
Hunt RD, Hartman RD, Molho-Pessach V, Votava HJ, Schaffer JV. Palisaded neutrophilic and granulomatous dermatitis in an adolescent girl with perinuclear antineutrophil cytoplasmic antibody-positive pauci-immune glomerulonephritis and arthritis. J Am Acad Dermatol. 2012;67(4):e164–6.
Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130(10):1278–83.
Ergun T, Inanc N, Tuney D, Kotiloglu EK, Seckin D, Tetik C, et al. Skin manifestations of rheumatoid arthritis: a study of 215 Turkish patients. Int J Dermatol. 2008;47(9):894–902.
Bremner R, Simpson E, White CR, Morrison L, Deodhar A. Palisaded neutrophilic and granulomatous dermatitis: an unusual cutaneous manifestation of immune-mediated disorders. Semin Arthritis Rheum. 2004;34(3):610–6.
Edgerton CC, Oglesby RJ, Bray D. Rheumatoid neutrophilic dermatitis. J Clin Rheum. 2006;12(5):266–7.
Yamamoto T, Ohkubo H, Nishioka K. Skin manifestations associated with rheumatoid arthritis. J Dermatol. 1995;22(5):324–9.
Bevin AA, Steger J, Mannino S. Rheumatoid neutrophilic dermatitis. Cutis. 2006;78(2):133–6.
Sanchez JL, Cruz A. Rheumatoid neutrophilic dermatitis. J Am Acad Dermatol. 1990;22(5 Pt 2):922–5.
Hirota TK, Keough GC, David-Bajar K, McCollough ML. Rheumatoid neutrophilic dermatitis. Cutis. 1997;60(4):203–5.
Setterfield JF, Shirlaw PJ, Challacombe SJ, Black MM. Pyoderma gangrenosum associated with severe oropharyngeal involvement and IgA paraproteinaemia. Br J Dermatol. 2001;144(2):393–6.
Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol. 2012;13(3):191–211.
Perry HO, Winkelmann RK. Bullous pyoderma gangrenosum and leukemia. Arch Dermatol. 1972;106(6):901–5.
O’Loughlin S, Perry HO. A diffuse pustular eruption associated with ulcerative colitis. Arch Dermatol. 1978;114(7):1061–4.
Su WP, Davis MD, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43(11):790–800.
Brooklyn TN, Dunnill MG, Shetty A, Bowden JJ, Williams JD, Griffiths CE, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55(4):505–9.
Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011;41(5):1203–17.
Hadi A, Lebwohl M. Clinical features of pyoderma gangrenosum and current diagnostic trends. J Am Acad Dermatol. 2011;64(5):950–4.
Weenig RH, Davis MD, Dahl PR, Su WP. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347(18):1412–8.
Acknowledgments
All authors contributed equally to the preparation of this review.
No sources of funding were used to prepare this review. A. Alavi, D. Sajic, F. B. Cerci, D. Ghazarian, M. Rosenbach, and J. Jorizzo have no conflicts of interest that are directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Alavi, A., Sajic, D., Cerci, F.B. et al. Neutrophilic Dermatoses: An Update. Am J Clin Dermatol 15, 413–423 (2014). https://doi.org/10.1007/s40257-014-0092-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-014-0092-6