
Abstract
Cutaneous small-vessel vasculitis (CSVV) is a disorder characterized by neutrophilic inflammation predominantly limited to the superficial cutaneous postcapillary venules. CSVV may be idiopathic or may have a defined cause such as infection, medication, connective tissue disease, or malignancy. CSVV may also be associated with extracutaneous disease or systemic vasculitis. The most common clinical presentation of CSVV consists of symmetrically distributed palpable purpura of the lower extremities. In general, lesional skin biopsy samples should be examined with light microscopy and direct immunofluorescence for adult patients with suspected CSVV. A complete history, review of systems, physical examination, and selected laboratory studies also should be performed to assess for inciting causes or extracutaneous involvement of CSVV. Treatment varies and depends on the chronicity of CSVV, the severity of cutaneous involvement, and the presence or absence of both an underlying cause and extracutaneous involvement of CSVV. An isolated episode of CSVV associated with a known inciting factor may be managed by removal or treatment of the trigger, along with symptomatic measures. First-line systemic treatments for chronic, idiopathic CSVV include colchicine or dapsone, used singly or in combination. Recurrent, chronic, or severely symptomatic CSVV that does not respond to the aforementioned therapies may require initiation of an immunosuppressive agent such as azathioprine, mycophenolate mofetil, methotrexate, cyclosporine, or rituximab.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cutaneous small-vessel vasculitis (CSVV) may be idiopathic or may have a defined cause such as infection, medication, autoimmune connective tissue disease, or malignancy |
The approach to a patient with suspected CSVV includes a careful evaluation of the cutaneous morphology, assessment of symptoms or signs indicating extracutaneous involvement, and identification of possible causes of CSVV |
Treatment of CSVV varies and depends on the chronicity of skin disease, underlying cause, presence or absence of extracutaneous involvement, and severity of cutaneous manifestations |
1 Introduction
Classification of vasculitis remains challenging. Two commonly used schemes are the American College of Rheumatology (ACR) classification criteria [1] and the Chapel Hill Consensus Conference (CHCC) nomenclature system [2]. The ACR and CHCC systems were intended as clinical research tools and a nomenclature system, respectively, rather than as diagnostic criteria for clinicians. A more useful classification scheme for the practicing dermatologist is one based on vessel-size predominance [2, 3].
Numerous terms have been used in the literature for vasculitides predominantly affecting the skin. The CHCC revised 2012 nomenclature system refers to this entity as cutaneous leukocytoclastic angiitis [2], whereas the ACR criteria of 1990 refer to this entity as hypersensitivity vasculitis [1]. Our preferred terminology is cutaneous small-vessel vasculitis (CSVV) [3, 4].
CSVV is primarily limited to the small blood vessels of the skin but may be associated with larger systemic vasculitis or extracutaneous involvement [5, 6]. Histologically, CSVV is characterized by perivascular neutrophilic inflammation of the postcapillary venules, fibrinoid destruction of the vessel walls, swelling of endothelial cells, and extravasation of red blood cells (i.e., leukocytoclastic vasculitis) [4]. When vasculitis is idiopathic (i.e., no evidence of infection, medication, or other identifiable cause), it is termed primary CSVV. Although idiopathic cases of CSVV do occur, they may be associated with drug hypersensitivities, systemic inflammatory conditions, connective tissue diseases, infections, and malignancies [4–8]. Various CSVV subtypes include immunoglobulin (Ig) A vasculitis (Henoch-Schönlein purpura [HSP]) [2], urticarial vasculitis, and cryoglobulinemic vasculitis. The purpose of this overview is to provide a practical guide to diagnosing, evaluating, and managing CSVV.
2 Clinical Features, Etiology, and Diagnosis of Cutaneous Small-Vessel Vasculitis
2.1 Clinical Features and Etiology
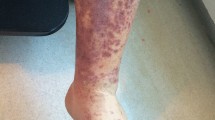
CSVV classically presents as symmetric palpable purpura of the lower extremities and other dependent areas of the body (Fig. 1). Lesions typically develop in crops and may be associated with pruritus, pain, and burning. Round, port wine-colored papules and plaques with inflammation may be seen [9]. Diascopy of purpuric lesions (application of direct pressure to the lesion with a glass slide) demonstrates partial blanching; the blanchable component indicates underlying inflammation (erythema), whereas the nonblanchable component represents hemorrhage (purpura) [9, 10]. Other clinical presentations include urticarial lesions [11], ulcerative or infarctive lesions, vesicles [12, 13], pustules, nodules, livedo pattern [14], and targetoid lesions [15, 16]. Ulcers or nodules may indicate deeper or medium-vessel involvement [17].

Clinical features of cutaneous small-vessel vasculitis. a Purple-red, partially blanchable and slightly elevated papules on the dorsal feet. b Coalescing palpable purpura on the lower extremities, with areas of ulceration. c Older lesions of palpable purpura in immunoglobulin A vasculitis, with areas showing a targetoid appearance
Although 45–55 % of CSVV is idiopathic [18], a search for underlying causes is essential. CSVV may be caused by infections (15–20 %), autoimmune connective tissue disease or inflammatory conditions (15–20 %), hypersensitivity drug reactions (10–15 %), or lymphoproliferative disorders or malignancies (5 %) [4, 18]. Specific causes of CSVV are listed in Table 1.
2.2 Diagnosis
2.2.1 Three Key Questions
Although histopathologic and laboratory tests are essential for the evaluation of CSVV, three key questions can be used to guide the clinician. First, is the morphology of the cutaneous eruption consistent with vasculitis? (Fig. 2a). CSVV commonly presents with palpable purpura developing in crops on dependent regions of the body. If the morphology is not consistent with CSVV, diagnoses such as thrombocytopenia, platelet dysfunction, and coagulation defects should be considered [19]. Second, does the patient have any symptoms or signs of extracutaneous vasculitis? This can be assessed by a detailed review of systems to screen for potential internal or systemic involvement (Fig. 2b). Third, does the patient have any identifiable causes for vasculitis or systemic disease associations? Underlying causes of vasculitis may be delineated with a complete history, review of systems, and physical examination, with particular emphasis on evaluation for new drug exposures, infectious conditions, autoimmune diseases, lymphoproliferative disorders, or malignancy (Table 1).

Approach to cutaneous small-vessel vasculitis. a Morphology. b Evaluation. ANCA anti-neutrophil cytoplasmic antibody, BMZ basement membrane zone, CTD connective tissue disease, DIF direct immunofluorescence, H&E hematoxylin and eosin, HSP Henoch-Schönlein purpura, Ig immunoglobulin, SLE systemic lupus erythematosus
2.2.2 Skin Biopsy
The diagnosis of vasculitis should typically be confirmed in adults with a skin biopsy. This usually includes a lesional specimen for light microscopy (hematoxylin and eosin [H&E]) and direct immunofluorescence (DIF) (Fig. 2a). Ideally, separate biopsy samples for H&E and DIF should be obtained. Histopathology of CSVV classically reveals a polymorphonuclear infiltrate, primarily affecting postcapillary venules, with fibrinoid deposits in and around the vessel wall, endothelial swelling, and extravasation of red blood cells (i.e., leukocytoclastic vasculitis) (Fig. 3) [4]. Notably, this histologic pattern is dynamic; in particular, the timing and location of the biopsy are critical for an accurate diagnosis. For H&E, a lesion that has been present for only 18–48 hours is ideal. After 48 hours, histopathology is usually nonspecific, showing an inflammatory reaction with a predominance of mononuclear cells, rather than neutrophils [4]. Additionally, it is imperative to obtain subcutaneous tissue in the biopsy specimen because deeper dermal involvement may be associated with systemic disease such as malignancy or connective tissue disease [20]. Reports of the severity of histopathologic changes predicting clinical severity are mixed [21, 22].

Histopathology of cutaneous small-vessel vasculitis. Perivascular neutrophilic inflammation of the postcapillary venules, fibrinoid destruction of the vessel walls, swelling of endothelial cells, and extravasation of red blood cells are consistent with leukocytoclastic vasculitis (hematoxylin and eosin; original magnification, ×10). (Photograph courtesy of Lawrence E. Gibson, MD.)
Cutaneous vasculitis has other histopathologic variants with associated underlying causes. For example, granulomatous vasculitis is more frequently associated with systemic vasculitis or systemic illness [23], whereas lymphocytic vasculitis may be associated with autoimmune connective tissue disease, viral infection, or drug reaction [4]. Additionally, some posit that lymphocytic vasculitis represents a reactive process rather than a primary disease [4, 24].
Other histologic findings may help determine the appropriate subtype of CSVV and identify its cause. CSVV involving small- and medium-sized vessels may suggest vasculitis associated with antineutrophil cytoplasmic antibodies or connective tissue disease [3]. In patients with urticarial lesions suggestive of urticarial vasculitis, a dermal interstitial neutrophilic infiltrate may indicate hypocomplementemic urticarial vasculitis, which is more commonly associated with systemic lupus erythematosus [4, 11, 25]. Tissue eosinophilia may indicate medication-induced CSVV [26]. Moreover, the absence of eosinophils can predict the risk of renal disease in adult patients with IgA vasculitis (HSP subtype), which may be a useful renal risk stratification tool when evaluating adults with HSP. The combination of leukocytoclastic vasculitis without eosinophils in patients older than 40 years with HSP has been associated with a 75 % rate of renal disease, a risk nearly three times greater than for other adult patients with HSP [27].
2.2.3 Direct Immunofluorescence
The ideal time frame for obtaining a biopsy for DIF is 8–24 hours after lesion onset because immune complexes may dissipate within 48 hours [4, 28]. The main role of DIF is to identify major immunoreactants. Lesions with IgA predominance suggest HSP and are associated with a higher risk of renal involvement (Fig. 2a). Although the association with cutaneous IgA deposition and renal involvement is not uniform and repeated urinalyses over time remain a better means of assessing renal involvement in CSVV, a DIF demonstrating positive IgA vascular fluorescence may prompt the clinician to more vigilantly monitor serial urinalyses (compared with DIF lacking IgA predominance). Additionally, increased severity of leukocytoclasis and absence of IgM deposits has been significantly associated with relapsing disease in adults with HSP [29]. Prominent IgM deposition may suggest cryoglobulinemia or rheumatoid vasculitis [30]. Hypocomplementemic urticarial vasculitis (HUV) will often show strong C3 immunofluorescence of the blood vessels and basement membrane zone. A continuous, strong, granular deposition of immunoreactants (C3 and immunoglobulins) along the basement membrane zone, termed the lupus band, is also frequently associated with HUV and a higher incidence of systemic lupus erythematosus [11]. A negative DIF in the setting of confirmed CSVV may increase clinical suspicion for a pauci-immune primary systemic vasculitis [13].
2.2.4 Laboratory Evaluation
The main goals of the laboratory evaluation are to search for an underlying cause and to exclude systemic involvement (Fig. 2b). The laboratory evaluations performed differ greatly, depending on whether the disease has a clear inciting factor or cause, shows systemic involvement, or is recurrent. Table 2 outlines our preferred laboratory approach. Furthermore, in patients with chronic or recurrent CSVV of unclear origin, we also suggest periodic clinical and laboratory evaluation to exclude an evolving systemic disorder as the underlying cause.
2.3 Management of CSVV
We recommend considering four key principles when determining the appropriate treatment of CSVV (Table 3). Treatment varies greatly, depending on the answers to these questions.
In a patient with a single episode of CSVV, the initial intervention aims to identify the cause of the vasculitis and remove or treat the trigger, which may or may not lead to resolution. Importantly, in up to 50 % of patients with CSVV, no underlying cause will be discovered [18]. CSVV is typically self-limiting (spontaneous resolution); therefore, conservative treatment is favored initially [4, 31]. Table 4 outlines our recommendations for management. In mild disease without evidence of systemic involvement or development of necrosis or ulceration, treatment should focus on symptomatic alleviation (Table 4) [4, 32]. A short course of prednisone (40–60 mg/day) tapered over 4–6 weeks can be considered if the disease is widespread or severely symptomatic [33]. For patients with recurrence upon tapering or withdrawal of systemic corticosteroids, the addition of a corticosteroid-sparing agent should be considered, rather than reinitiation of corticosteroids [34].
For patients with chronic and refractory vasculitis or severe cutaneous disease, initiation of systemic therapies is indicated. Several reports [32, 35, 36] describe colchicine treatment as beneficial within 1–2 weeks; however, the only randomized controlled study to date for colchicine use in vasculitis did not demonstrate efficacy [37]. Although dapsone has been shown to be efficacious in case series [38–41], no placebo-controlled trials have been performed.
Second-line treatments for recurrent, chronic, or severely symptomatic CSVV include immunosuppressive agents. We prefer to start with mycophenolate mofetil (given its relative ease of laboratory monitoring and favorable adverse-effect profile compared with other immunosuppressive agents), which was recently reported to be effective in a patient with ulcerative CSVV recalcitrant to colchicine and dapsone [42]. Earlier studies have also supported its use in systemic necrotizing vasculitis and HUV [4, 43]. When treating with azathioprine, it is recommended to titrate the medication dosage according to the thiopurine methyltransferase enzyme levels [4]. If CSVV is recalcitrant to both mycophenolate mofetil and azathioprine, methotrexate can be considered. Although methotrexate has shown reasonable efficacy, it is important to be aware that methotrexate-induced CSVV has been reported [4]. Importantly, treatment recommendations can be modified, depending on factors such as patient preferences or comorbid conditions.
Many other treatment options are available for refractory, idiopathic CSVV; we do not routinely use them, although some have been reported as moderately successful in case reports or series. Elimination diets showed improvement in two case series [44, 45]. Hydroxychloroquine has successfully treated HUV [46], and rituximab has been useful in idiopathic CSVV [47]. Tumor necrosis factor–α antagonists have also been used to treat vasculitis, but we generally avoid them out of concern for potentially precipitating CSVV [48]. Moreover, these medications may mask the cause of CSVV for patients with systemic diseases (e.g., rheumatoid arthritis, inflammatory bowel disease) because CSVV may be caused by the biologic medication or by the systemic disease [48–50].
Patients with persistent ulceration or common variable immunodeficiency associated with CSVV may benefit from treatment with intravenous immunoglobulin [51, 52]. Plasmapheresis has also been used to treat patients with refractory CSVV [53]. Additional immunosuppressants (e.g., cyclophosphamide, cyclosporine) are not commonly used but may be beneficial [5, 54].
3 Conclusion
Table 5 summarizes key points for the diagnosis, evaluation, and management of CSVV. Clinicians should be aware of the clinical and morphologic presentations of CSVV and the importance of determining extracutaneous involvement and identifiable causes of disease. Many treatment options are available, and therapeutic decisions depend on numerous factors such as whether the episode of vasculitis is isolated or recurrent, has an identifiable cause, affects the patient systemically, or has severe cutaneous involvement. The summary of our approach described here provides helpful information to clinicians encountering patients with CSVV in the outpatient and hospital settings.
References
Hunder GG, Arend WP, Bloch DA, Calabrese LH, Fauci AS, Fries JF, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: introduction. Arthritis Rheum. 1990;33(8):1065–7.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
Fiorentino DF. Cutaneous vasculitis. J Am Acad Dermatol. 2003;48(3):311–40.
Russell JP, Gibson LE. Primary cutaneous small vessel vasculitis: approach to diagnosis and treatment. Int J Dermatol. 2006;45(1):3–13.
Marzano AV, Vezzoli P, Berti E. Skin involvement in cutaneous and systemic vasculitis. Autoimmun Rev. 2013;12(4):467–76 (Epub 2012 Aug 16).
Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M. Cutaneous vasculitis in children and adults: associated diseases and etiologic factors in 303 patients. Medicine (Baltimore). 1998;77(6):403–18.
Martinez-Taboada VM, Blanco R, Garcia-Fuentes M, Rodriguez-Valverde V. Clinical features and outcome of 95 patients with hypersensitivity vasculitis. Am J Med. 1997;102(2):186–91.
Loricera J, Calvo-Rio V, Ortiz-Sanjuan F, Gonzalez-Lopez MA, Fernandez-Llaca H, Rueda-Gotor J, et al. The spectrum of paraneoplastic cutaneous vasculitis in a defined population: incidence and clinical features. Medicine (Baltimore). 2013;92(6):331–43.
Piette WW. The differential diagnosis of purpura from a morphologic perspective. Adv Dermatol. 1994;9:3–23.
Piette WW. Purpura. In Callen JP, Jorizzo JL, Bolognia JL, Piette WW, Zone JJ, editors. Dermatological signs of internal disease. 4th ed. London: Saunders/Elsevier; 2009. p. 85–92.
Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol. 1992;26(3 Pt 2):441–8.
Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology. 2010;56(1):3–23.
Fett N. Evaluation of adults with cutaneous lesions of vasculitis [Internet]. Topic 13769 Version 11.0. UpToDate; 2014 [updated 2013 Oct 10; cited 2014 Jan 16]. http://www.uptodate.com/contents/evaluation-of-adults-with-cutaneous-lesions-of-vasculitis.
Dhadly M, Dean SM, Eberhardt RT. Cutaneous changes in peripheral vascular arterial disease. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, editors. Fitzpatrick’s dermatology in general medicine. 7th ed. Vol. 2. New York (NY): McGraw-Hill Medical; 2008. p. 1667–79.
Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51(8):889–902.
Hughey LC. Approach to the hospitalized patient with targetoid lesions. Dermatol Ther. 2011;24(2):196–206.
Xu LY, Esparza EM, Anadkat MJ, Crone KG, Brasington RD. Cutaneous manifestations of vasculitis. Semin Arthritis Rheum. 2009;38(5):348–60 Epub 2008 Mar 20.
Chung L, Kea B, Fiorentino DF. Cutaneous vasculitis. In: Bolognia JL, Jorizzo JL, Rapini RD, editors. Dermatology. 2nd ed. Vol. 1. St. Louis (MO): Mosby Elsevier; 2008. p. 347–67.
Wetter DA. Purpura. In: Mattuci-Cerinic M, Furst D, Fiorentino DF, editors. Skin manifestations of rheumatic disease. New York (NY): Springer; 2013. p. 47–54.
Sanchez NP, Van Hale HM, Su WP. Clinical and histopathologic spectrum of necrotizing vasculitis: report of findings in 101 cases. Arch Dermatol. 1985;121(2):220–4.
Hodge SJ, Callen JP, Ekenstam E. Cutaneous leukocytoclastic vasculitis: correlation of histopathological changes with clinical severity and course. J Cutan Pathol. 1987;14(5):279–84.
Cribier B, Couilliet D, Meyer P, Grosshans E. The severity of histopathological changes of leukocytoclastic vasculitis is not predictive of extracutaneous involvement. Am J Dermatopathol. 1999;21(6):532–6.
Gibson LE, Winkelmann RK. Cutaneous granulomatous vasculitis: its relationship to systemic disease. J Am Acad Dermatol. 1986;14(3):492–501.
Massa MC, Su WP. Lymphocytic vasculitis: is it a specific clinicopathologic entity? J Cutan Pathol. 1984;11(2):132–9.
Davis MD, Daoud MS, Kirby B, Gibson LE, Rogers RS 3rd. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Dermatol. 1998;38(6 Pt 1):899–905.
Bahrami S, Malone JC, Webb KG, Callen JP. Tissue eosinophilia as an indicator of drug-induced cutaneous small-vessel vasculitis. Arch Dermatol. 2006;142(2):155–61.
Poterucha TJ, Wetter DA, Gibson LE, Camilleri MJ, Lohse CM. Histopathology and correlates of systemic disease in adult Henoch-Schönlein purpura: a retrospective study of microscopic and clinical findings in 68 patients at Mayo Clinic. J Am Acad Dermatol. 2013;68(3):420–4.e3 (Epub 2012 Sep 6).
Schroeter AL, Copeman PW, Jordon RE, Sams WM Jr, Winkelmann RK. Immunofluorescence of cutaneous vasculitis associated with systemic disease. Arch Dermatol. 1971;103(3):254–9.
Byun JW, Song HJ, Kim L, Shin JH, Choi GS. Predictive factors of relapse in adult with Henoch-Schönlein purpura. Am J Dermatopathol. 2012;34(2):139–44.
Gibson LE. Cutaneous vasculitis update. Dermatol Clin. 2001;19(4):603–15.
Callen JP. A clinical approach to the vasculitis patient in the dermatologic office. Clin Dermatol. 1999;17(5):549–53.
Kinney MA, Jorizzo JL. Small-vessel vasculitis. Dermatol Ther. 2012;25(2):148–57.
Lotti T, Ghersetich I, Comacchi C, Jorizzo JL. Cutaneous small-vessel vasculitis. J Am Acad Dermatol. 1998;39(5 Pt 1):667–87.
Fett N. Management of adults with idiopathic cutaneous small vessel vasculitis [Internet]. Topic 13785 Version 5.0. UpToDate; 2014 [updated 2013 Sep 6; cited 2014 Jan 16]. http://www.uptodate.com/contents/management-of-adults-with-idiopathic-cutaneous-small-vessel-vasculitis.
Callen JP. Colchicine is effective in controlling chronic cutaneous leukocytoclastic vasculitis. J Am Acad Dermatol. 1985;13(2 Pt 1):193–200.
Asherson RA, Buchanan N, Kenwright S, Fletcher CM, Hughes GR. The normocomplementemic urticarial vasculitis syndrome: report of a case and response to colchicine. Clin Exp Dermatol. 1991;16(6):424–7.
Sais G, Vidaller A, Jucgla A, Gallardo F, Peyri J. Colchicine in the treatment of cutaneous leukocytoclastic vasculitis: results of a prospective, randomized controlled trial. Arch Dermatol. 1995;131(12):1399–402.
Swerlick RA, Lawley TJ. Small-vessel vasculitis and cutaneous vasculitis. In: Churg A, Churg J, editors. Systemic vasculitides. New York (NY): Igaku-Shoin Medical Publishers, Inc; 1991. p. 193–201.
Ruiz Villaverde R, Blasco Melguizo J, Martin Sanchez MC, Naranjo Sintes R. Annular leucocytoclastic vasculitis: response to dapsone. J Eur Acad Dermatol Venereol. 2002;16(5):544–6.
Fredenberg MF, Malkinson FD. Sulfone therapy in the treatment of leukocytoclastic vasculitis: report of three cases. J Am Acad Dermatol. 1987;16(4):772–8.
Fortson JS, Zone JJ, Hammond ME, Groggel GC. Hypocomplementemic urticarial vasculitis syndrome responsive to dapsone. J Am Acad Dermatol. 1986;15(5 Pt 2):1137–42.
Haeberle MT, Adams WB, Callen JP. Treatment of severe cutaneous small-vessel vasculitis with mycophenolate mofetil. Arch Dermatol. 2012;148(8):887–8.
Worm M, Sterry W, Kolde G. Mycophenolate mofetil is effective for maintenance therapy of hypocomplementaemic urticarial vasculitis. Br J Dermatol. 2000;143(6):1324.
Lunardi C, Bambara LM, Biasi D, Zagni P, Caramaschi P, Pacor ML. Elimination diet in the treatment of selected patients with hypersensitivity vasculitis. Clin Exp Rheumatol. 1992;10(2):131–5.
Ferri C, Pietrogrande M, Cecchetti R, Tavoni A, Cefalo A, Buzzetti G, et al. Low-antigen-content diet in the treatment of patients with mixed cryoglobulinemia. Am J Med. 1989;87(5):519–24.
Lopez LR, Davis KC, Kohler PF, Schocket AL. The hypocomplementemic urticarial-vasculitis syndrome: therapeutic response to hydroxychloroquine. J Allergy Clin Immunol. 1984;73(5 Pt 1):600–3.
Chung L, Funke AA, Chakravarty EF, Callen JP, Fiorentino DF. Successful use of rituximab for cutaneous vasculitis. Arch Dermatol. 2006;142(11):1407–10.
Sokumbi O, Wetter DA, Makol A, Warrington KJ. Vasculitis associated with tumor necrosis factor-α inhibitors. Mayo Clin Proc. 2012;87(8):739–45 Epub 2012 Jul 13.
Marzano AV, Borghi A, Stadnicki A, Crosti C, Cugno M. Cutaneous manifestations in patients with inflammatory bowel diseases: pathophysiology, clinical features, and therapy. Inflamm Bowel Dis. 2014;20(1):213–27.
Marzano AV, Borghi A, Meroni PL, Crosti C, Cugno M. Immune-mediated inflammatory reactions and tumors as skin side effects of inflammatory bowel disease therapy. Autoimmunity. 2014 Jan 20 [Epub ahead of print].
Ong CS, Benson EM. Successful treatment of chronic leukocytoclastic vasculitis and persistent ulceration with intravenous immunoglobulin. Br J Dermatol. 2000;143(2):447–9.
Sais G, Vidaller A, Servitje O, Jucgla A, Peyrí J. Leukocytoclastic vasculitis and common variable immunodeficiency: successful treatment with intravenous immune globulin. J Allergy Clin Immunol. 1996;98(1):232–3.
Turner AN, Whittaker S, Banks I, Jones RR, Pusey CD. Plasma exchange in refractory cutaneous vasculitis. Br J Dermatol. 1990;122(3):411–5.
Nett N, Callen JP. Leukocytoclastic vasculitis. In: Lebwohl MG, Heymann WR, Berth-Jones J, Coulson I, editors. Treatment of skin disease: comprehensive therapeutic strategies. 4th ed. Edinburgh: Elsevier/Saunders; 2014. p. 384–7.
Acknowledgements
No sources of funding were used to prepare this review. M. R. Goeser, V. Laniosz, and D. A. Wetter have no conflicts of interest that are directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Goeser, M.R., Laniosz, V. & Wetter, D.A. A Practical Approach to the Diagnosis, Evaluation, and Management of Cutaneous Small-Vessel Vasculitis. Am J Clin Dermatol 15, 299–306 (2014). https://doi.org/10.1007/s40257-014-0076-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-014-0076-6