
Abstract
Laser sintering (LS) of polyamide 12 (PA12) is increasingly being adopted for industrial production of end-use parts, yet the complexity of this process coupled with the lack of organized, rigorous, publicly available process-structure-physical property datasets exposes manufacturers and customers to risks of unacceptably poor part quality and high costs. Although an extensive scientific literature has been developed to address some of these concerns, results are distributed among numerous reports based on different machines, materials, process parameters, and users. In this study, a single commercially important LS PA12 feedstock has been processed along four build dimensions of a modern production LS machine, characterized by a wide range of physical techniques, and compared to the same material formed by conventional melt processing. Results are discussed in the context of the literature, offering novel insights including distributions of particle size and shape, localization of semicrystalline phase changes due to LS processing, effect of chemical aging on melt viscosity, porosity orientation relative to LS build axes, and microstructural effects on tensile properties and failure mechanisms. The resulting datasets will be made publicly available to modelers and practitioners for the purpose of improving certifiability and repeatability of end-use parts manufactured by LS.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Additive manufacturing (AM) technologies are increasingly being adopted by corporations, small businesses, militaries, governments, and individuals for low- to medium-volume production of end-use products [1,2,3,4]. However, the roots of these technologies in rapid prototyping (RP) have resulted in limitations for true manufacturing purposes, including a lack of repeatability leading to uncertainty in whether parts will meet quality control requirements. This situation arises from a number of both physical and economic challenges. Physical challenges include the complexity of AM processes and resulting dependence of part properties on their location in the build envelope, temperature control throughout the nonisothermal and dynamic process, part dimensions and geometry, part orientation with respect to machine axes and toolpath, and material condition (fresh vs. recycled). Economic challenges include the high cost of many AM machines, materials, workforce training, and labor, as well as the proprietary nature of some AM materials and processes, leading to a lack of a public knowledge base. Predictive simulations can in principle streamline manufacturing workflows to mitigate risks and reduce costs. Yet the above challenges have thus far impeded the organized collection of rigorous, publicly available datasets that could serve as input parameters for simulations as well as guidelines for standards and certification of processes and parts. The Additive Manufacturing Benchmarks (AM Bench) initiative aims to address this need by establishing a continuing series of measurement challenges aimed at validating and improving models and developing universally accepted measurement approaches for AM.
Polymer powder bed fusion (PPBF) is one of the most established AM methods and was a leading technology in the transition from RP to AM in the 1990s and 2000s [5,6,7,8]. The most common PPBF type is laser sintering (LS), developed in the 1980s at the University of Texas and commercialized first by desktop manufacturing (DTM), then later 3D Systems, Electro Optical Systems (EOS), Farsoon, and others. LS is currently used for prototyping as well as production in numerous industries including aerospace and defense [9,10,11,12,13], transportation [14–15], prosthetics and orthotics [16,17,18,19,20], industrial equipment [21], electronics [22–23], sporting goods [24,25,26,27], and fashion [28]. LS is well suited to batchwise production of many nested components and assemblies up to 100’s of millimeters in size within a single build. The self-supporting nature of the powder bed makes it one of the most attractive AM technologies for producing lightweight, complex parts and assemblies with overhangs, lattice structures, integrated hinges, snap clips and other joints, interior baffles for ducts and fuel tanks, coil or leaf springs, chains, screw threads, and designed failure paths [29]. The majority of commercial LS feedstocks are based on polyamide 12 (PA12), which is uniquely suited to the LS process and tends to reliably produce parts with good consolidation and dimensional accuracy while offering superior thermomechanical properties and chemical resistance to materials used in most other polymer AM technologies. However, parts made by LS typically possess reduced ductility and fracture resistance relative to conventionally melt-processed PA12, as well as moderate anisotropy in the through-layer (Z) dimension and a rough surface texture that may need to be removed by media tumbling for some applications [30].
Figure 1 provides an overview of a common production LS machine, the ProX500 from 3D SystemsFootnote 1 Unlike in the older DTM design, the powder feedstock is fed from above the build platform, enabling continuous feed refreshment during builds and recycling of overflow powder from layer deposition. A counter-rotating roller applicator deposits powder layers on the order of 100 μm thick, using a return piston to distribute powder over both the forward and return strokes in an attempt to achieve greater densification than the original single-pass design [31]. Each freshly applied layer is preheated by an eight-zone array of quartz lamps above the bed for about 5 s until it reaches a temperature setpoint just below its melting point. Nominally 2D slices of the parts are then formed by rastering a CO2 laser over selected regions of the layer, taking between 5 s and 40 s to melt and fuse the powder depending on the number and size of the slices. The build plate then moves down a distance which sets the layer thickness (typically 100 μm), and the process is repeated until the parts are completed.

(a) Process schematic for 3D Systems ProX500 production laser sintering (LS) machine. (b) Stepwise schematic of powder layer deposition, preheating, and laser fusion
Polyamides (nylons) are important engineering thermoplastics used in automotive, medical, food packaging, electronics, textiles, and other industries. Among nylons, PA12 has some particularly desirable properties including low water absorption, high resistance to fracture from impact or fatigue including at low temperatures, good sliding friction and abrasion resistance, low density, and low processing temperature [32–33]. These properties arise from its relatively long polyethylene-like aliphatic segments (Fig. 2a) separated by hydrogen-bonding amide groups that template the semicrystalline microstructure (Fig. 2b, c) and impart higher melting point, glass transition temperature, stiffness, and strength than commodity polyolefins. Similar to other even-numbered n-nylons such as PA6 [34–35], PA12 can adopt two primary crystal structures: the α phase in which antiparallel chains form hydrogen-bonded sheets with a monoclinic unit cell, or the γ phase in which amide groups twist approximately 60° out of the CH2 zigzag plane to form parallel-oriented hydrogen-bonded sheets on a pseudohexagonal lattice [36–37]. Schematics of chain stem packing in α and γ phases are shown in Fig. 2b, c, respectively. In PA12, the γ form is favored due to the ability of the relatively long CH2 segments to relax the torsional stress of this twisting [32].

a Scheme of thermally driven condensation reaction between unprotected amine and carboxylic acid end groups of PA12, resulting in increased polymer molecular mass. Chain stem packing in b α and c γ phase crystal structures of PA12. Solid circles represent chains pointing up; open circles are chains pointing down. Dashed lines represent hydrogen bonds. Adapted from [32]
PA12 is industrially synthesized by ring opening polymerization of lauryl lactam, resulting in one amine and one carboxylic acid end group. These end groups are typically protected in an additional reaction step to avoid molecular mass increase during processing due to a thermally driven condensation reaction, shown in Fig. 2a. However, for LS grades of PA12, the end groups are often left unprotected in order to leverage this reaction for balancing the needs of low initial zero shear viscosity with good layer adhesion and part mechanical properties [38]. Polyamides are conventionally melt processed by techniques such as extrusion, injection molding, and compression molding. In this work, LS grade PA12 has been compression molded as a representative baseline for comparing properties of melt-processed PA12 to samples formed by LS from the same feedstock.
Several LS benchmark studies have been published [39,41,42,43,43] that investigate challenging part geometries to determine optimum build parameters for feature resolution and accuracy. In this work, we focus on in-depth characterization of physical properties using standard test specimens formed along multiple build axes using the LS machine manufacturer’s recommended processing conditions, which aim for a balance between feature resolution and mechanical performance. The ProX500Footnote 2 is chosen as a representative example of current production LS technology. This machine is widely used in industry due to its automated powder handling and dual-pass applicator design (Fig. 1), whereas the scientific LS literature has primarily investigated older machine designs that may not generate comparable part properties. Many authors have characterized processing-microstructure-property relationships for LS PA12 in great detail; however, the results are distributed among numerous papers using a wide variety of PA12 feedstocks, machines, and operators, making it difficult to draw general conclusions. This study aims to provide a thorough, organized dataset that is fundamentally rigorous and useful for modelers, practitioners, and standards organizations alike in the continued development of LS technology towards manufacturing end-use products with certifiable and repeatable performance. In addition, the results have been analyzed in the context of scientific knowledge on polyamides as well as the LS process. Raw data files for all of the results presented here will be made available free of charge at the AM Bench website (https://www.nist.gov/ambench).
Experimental
Materials
Duraform ProX PA powder was obtained from 3D Systems (Rock Hill, SC, USA). Trifluoroacetic anhydride (TFAA), dichloromethane (DCM), and dimethylformamide (DMF) were obtained from Sigma-Aldrich and used as received.
Scanning Electron Microscopy
Virgin Duraform powder was sprinkled onto a piece of double-sided carbon tape affixed to a scanning electron microscopy (SEM) sample holder, and the specimen was blasted with compressed nitrogen to remove loosely affixed particles. Fracture surfaces from tensile specimens were mounted directly onto carbon tape on similar holders. Specimens were sputtered with an approximately 10-nm-thick film of gold-palladium and imaged in an S-4700 scanning electron microscope (Hitachi Ltd., Tokyo, Japan) using an accelerating voltage of 5 kV.
Camsizer Particle Size Distribution Analysis
Approximately 1-mL samples of powder were fed by a vibrating tray into a Camsizer X2 optical particle analyzer (Retsch Technology GmbH, Haan, Germany). Inside the Camsizer, powder was fluidized by nitrogen gas at 50 kPa through a 4-mm nozzle, forming a continuous jet that passed through an optical chamber illuminated by a pair of pulsed LEDs. 2D images of particles were recorded by a pair of CCD cameras with resolutions of 0.8–50 μm (zoom camera) and 50 μm to 8 mm (base camera). The Camsizer software tracks and processes a number of individual particles from each image, using their projected area and perimeter to calculate a range of size and shape parameters, some of which are discussed in the text in “Particle Size and Shape Distributions.” Reported distributions from Camsizer measurements represent the average of three runs of the virgin Duraform powder. During each run, between 300,000 and 1,000,000 individual particles were imaged and analyzed over the course of 30–70 s per run.
Size Exclusion Chromatography
PA12 is insoluble in most common solvents but can be rendered soluble by reaction with TFAA [44–45]. One hundred milligrams (approximately 0.5 mmol of amide groups) of virgin Duraform ProX powder was added to 3 mL anhydrous DCM in a dried 50 mL round-bottom flask. With vigorous stirring, 1 mL (1.51 g, 7.2 mmol) of TFAA was added. The white powder quickly agglomerated and dissolved within 1 min to form a transparent solution. The flask was sealed under nitrogen gas and stirred at room temperature for 21 h, then opened. It was purged with nitrogen gas for 20 min causing evaporation of DCM, leaving a transparent viscous liquid product. Fifty milliliters of DMF containing a small amount of toluene as a flow marker for size exclusion chromatography (SEC) was added, and the flask was agitated on a wrist-action shaker for 2 h to dissolve the product. The solution was filtered through a 0.2-μm PTFE syringe filter and 100 μL was injected into a 1260 Infinity HPLC system (Agilent Technologies, Santa Clara, CA, USA) pumping DMF at 0.7 mL/min through two Agilent PLGel 5 μm mixed D SEC columns followed by an Optilab rEX differential refractive index (dRI) detector (Wyatt Technology, Santa Barbara, CA, USA). Relative molecular weight was determined using conventional calibration with Viscotek PolyCAL polystyrene standards (Spectris plc, Surrey, UK) covering a range from 1 to 170 kDa.
Laser Sintering
100% virgin Duraform ProX PA powder was fed automatically from a 3D Systems material quality control (MQC) system into a 3D Systems ProX500 laser sintering machine. The process chamber environment was held below 5% O2 via continuous nitrogen gas purge. The default processing conditions for the material were used, including a 105 °C setpoint for the hopper feed module (which is expected to provide a moderate amount of powder drying during the warmup and build phases), layer thickness of 100 μm, laser speed vl of 12 m/s, laser scan spacing ds of 200 μm, fill laser power Pl of 62 W, and outline laser power of 20 W. This yields a laser energy density De of approximately 2.6 J/cm2 according to the well-known formula
The warmup phase lasted 1 h 11 min during which a 13-mm (130 layers) base of unfused powder was deposited while heating from an initial bed temperature of 130 °C. At 4.57 mm, the bed temperature setpoint was increased to 155 °C and at 6.35 mm, it was increased to 169 °C where it was held for the remainder of warmup and into the build phase. The build phase lasted 6 h 40 min during which 118 mm (1180 layers) were applied and selected regions were fused by the laser. The build design is shown in Fig. 3, featuring arrays of ASTM D638 type IV tensile specimens oriented along various axes of the LS machine as defined in ISO/ASTM 52921 [46]. The part orientation labeling scheme is described in the caption of Fig. 3 and follows the same convention as Usher et al. [47] Parts were scanned by the laser in the same order for each layer, following the sequence of specimen numbers listed in Fig. 3 rather than allowing the software to dynamically choose the scan order. Raster direction was alternated layer by layer between X and Y directions (“Sinterscan” option which is the default for this system).

Build design for laser sintering (LS) specimens characterized in this work, featuring ASTM D638 type IV specimens oriented along various machine axes, identified by the orientation of the primary (tensile) axis followed by the orientation of the specimen width (broadest lateral dimension): samples 1–5 are identified as ZY oriented, samples 6–10 as YX, samples 11–15 as YZ, and samples 26–30 as 45° ZY
At a build height of 30 mm, the bed temperature setpoint was lowered from 169 to 167.5 °C, to account for excess thermal energy stored in the powder bed as recommended by the machine manufacturer. During the cooling phase, an additional 2.6 mm of powder (26 layers) was added on top of the completed build, followed by an uncontrolled cool down during which the bed surface temperature dropped at approximately 0.9 °C/min. The powder cake was left in the machine for 14 h before removing and breaking out the fused parts. Parts were finished by brushing and blasting with compressed air to remove loose powder.
Compression Molding and Machining
One hundred fifteen grams of virgin Duraform powder were dried in a vacuum oven at 75 °C for 15 h, then sandwiched between PTFE-impregnated fabric sheets on steel plates separated by a 3.175-mm-thick, 127-mm square steel mold coated with Frekote 55-NC mold release (Henkel AG, Düsseldorf, Germany). This assembly was held between the platens of a P300 P/M programmable melt press (Collin GmbH, Maitenbeth, Germany) at 230 °C for 4 min. A degassing cycle was repeated twice in which 15 bar of pressure was applied by the platens for 30 s, then the press was opened slightly for 15 s. Following this, 100 bar (10 MPa) of pressure was applied for 2 min, after which pressure was maintained at 10 MPa for another 40 min while temperature was decreased at a controlled rate of 5 °C/min. When the platens reached 30 °C, pressure was released and the solid PA12 sheet was removed. The sheet width was approximately 1 mm smaller than the mold cavity on all sides due to shrinkage. The sheet was polished on a Spectrum System 2000 rotary polisher (Leco Corporation, Saint Joseph, MI, USA) using 100MX and 240MX grit abrasive cloth (Micro-Surface Finishing Products, Wilton, IA, USA) at 180 rpm under a continuous stream of deionized water, then dried with paper towels. The 3.175-mm-thick sheet was machined into six ASTM D638 type IV tensile specimens using a DSLS 3000 Micro CNC Mill (Micro Proto Systems, Chandler, AZ USA). Specimens were sanded to deburr and dried in a vacuum oven at 75 °C for 1.5 h before testing. A second sheet was made by a similar procedure using a 1-mm-thick 250-mm square mold, but was not polished.
Thermogravimetric Analysis
Four milligrams of virgin Duraform powder was heated in a Q5000 thermogravimetric analyzer (TGA) (TA Instruments, New Castle, DE USA) in a high-temperature platinum pan under nitrogen purge at 20 °C/min from room temperature to 120 °C, held for 30 min at 120 °C to drive off moisture, then heated at 20 °C/min to 900 °C.
Dynamic Mechanical Analysis
Following testing, the grip section of LS tensile specimen 9 (Fig. 3) was cut into a 35 mm × 12 mm rectangle and milled to a thickness of 1.3 mm using a DSLS 3000 Micro CNC Mill. It was loaded in single cantilever mode on a TA Instruments Q800 dynamic mechanical analyzer (DMA) and tested using an oscillatory strain of 0.02% at a frequency of 1 Hz while ramping temperature at 1 °C/min from − 140 to 200 °C. The same method was used for two 35 mm × 12 mm rectangles cut from the 1-mm-thick melt-pressed film: one sample that was dried in a vacuum oven at 75 °C for 1.5 h before testing, and a second sample that was stored at ambient conditions (47% ± 11% relative humidity, 21 °C ± 4 °C) for 3 days before testing.
Differential Scanning Calorimetry
Three milligrams to 5 mg samples of virgin Duraform powder, a section cut from the grip section of LS tensile specimen 6 (Fig. 3) including the outer surface, and a section from the 1-mm-thick melt-pressed sheet were tested in a TA Instruments Discovery differential scanning calorimetry (DSC) using a cycle from room temperature to 225 °C, then down to − 90 °C, back up to 225 °C, and back down to − 90 °C at a constant rate of 10 °C/min.
X-Ray Diffraction
In a Bruker D8 Discover diffractometer equipped with a Cu Kα X-ray source (λ = 0.154 nm) operating at 40 kV and 40 mA, a Göbel mirror monochromator, and a LYNXEYE XE compound silicon strip 1D detector, samples were scanned over a range of diffraction angles 2Θ from 3° to 50° relative to the flat surface of grip sections of tested tensile specimens, or the virgin Duraform powder, using a theta-theta geometry (incident angle = diffracted angle = Θ). Full scans in ambient atmosphere with a step size of 0.050° took approximately 40 s total due to the high-throughput detector.
Fourier Transform Infrared and Raman Spectroscopy
Attenuated total reflectance (ATR) Fourier transform infrared (FTIR) spectra were recorded on a Thermo-Nicolet iS50 spectrometer with built-in diamond ATR crystal, using 32 scans per sample at a resolution of 2 cm−1. Fourier transform Raman spectra were recorded on the same spectrometer using the iS50 Raman attachment with excitation via a 1064-nm diode laser, using 2048 scans per sample at a resolution of 8 cm−1.
Melt Rheology
In an ARES G2 rheometer, 25-mm parallel plates were equilibrated at 200 °C for 30 min then zeroed. Virgin Duraform powder was loaded via a melt preparatory kit and the upper plate was closed to a normal force of 10 N ± 1 N, resulting in a gap of approximately 5 mm. Temperature was lowered to 80 °C and held for 1.5 h to dry the polymer, then the system was heated to 200 °C and the gap was closed to approximately 2 mm with 15 N force. The sample was trimmed and the gap closed to optimal height. Note the sample was held at 200 °C for approximately 9 min during the gap closure to 2 mm and trimming, during which time chemical aging (Fig. 2a) likely occurred prior to beginning experiments.
In the first experiment, angular frequency ω was ramped from 10−1 rad/s to 102 rad/s over 13 min with constant oscillatory strain of 10%, followed by a decreasing frequency ramp from 102 rad/s to 10−1 rad/s lasting another 13 min. In a second experiment, a fresh batch of Duraform was loaded as described above and subjected to a frequency sweep from 100 rad/s to 102 rad/s lasting 70 to 80 s at constant oscillatory strain of 3%, followed by a time sweep at constant frequency of 10 rad/s for 120 min. This sequence was repeated eight times for a total aging time of 16 h at 200 °C. Instrument error is ± 5% in frequency and ± 8% in modulus based on manufacturer specification.
Pycnometry
Samples weighing between 0.5 and 1 g were loaded into the small sample chamber of a Quantachrome Ultrapyc 1200e helium pycnometer (Anton Paar, Graz, Austria) which was calibrated using a standard steel sphere to an accuracy of ± 0.002 cm3. Volume was calculated from the average of five runs with a maximum standard deviation of 0.0031 cm3. Samples were weighed on a Cubis MSE225P-100DU semi-micro balance (Sartorius GmbH, Goettingen, Germany) with a resolution of ± 0.01 mg. The sample chamber volume (nominally 10.8 cm3) was only filled approximately 10% full, which is expected to introduce a maximum uncertainty of ± 1% to the volume determination [48].
X-Ray Micro-computed Tomography
Tensile specimens 3, 8, 13, and 28 (Fig. 3) were not tested mechanically; their gauge sections (3.5 mm × 6 mm × 30 mm rectangles) were cut out and imaged in a Zeiss Versa XRM-500 X-ray micro-computed tomography (X-μCT) machine at 60 kV and 5 W with a voxel size of 7.5 μm. Image Pro software was used to threshold images resulting in a 3D binary data structure in which pores are white and solid material is black. The 3D pores were then analyzed for volume, shape, and orientation parameters including length (L), defined as the longest vector that can be drawn from one point on the edge of a pore to any other, and width (W) and thickness (T), which are defined similarly to L but satisfying the conditions L ⊥ W ⊥ T, which implies that L ≥ W ≥ T. Ratios of L, W, and T then serve as aspect ratios approximately defining the pore shape [49].
Tensile Testing
Gauge sections of D638 type IV specimens were decorated with a fine speckle pattern using white and black spray paint for strain measurement via digital image correlation (DIC). After air drying for 1 h, they were tested in tension at a constant displacement rate of 5 mm/min on an Instron model 5985 frame (Illinois Tool Works, Glenview, IL, USA) with 10 kN load cell using pneumatically actuated serrated grips. DIC images were recorded at 1 frame/s using the frame’s built-in camera and an AF-D NIKKOR 24–85 mm f/1:2.8–4 lens (Nikon Corporation, Tokyo, Japan) and correlated using Instron Bluehill software with a subset size of 31 pixels.
Results and Discussion
Particle Size and Shape Distributions
Particle shape and size distribution are critical parameters for LS due to their effect on powder flowability and resulting layer smoothness, uniformity, and density [38, 50]. Figure 4a shows a scanning electron micrograph (SEM) of virgin Duraform ProX polyamide (PA) powder. The particles have a “potato”-like shape resulting from their formation via precipitation from ethanol under pressure [51–52]. This type of powder has been shown to flow and pack reasonably well in LS with the addition of hydrophobically modified fumed silica as a flow-enhancing agent [53]. Duraform powder originates from the same manufacturer as the common PA2200 PA12 powder from EOS, but their particle size distributions are different [38]. Historically, Duraform powder tends to have a broader size distribution with a higher fraction of small particles than PA2200, favoring packing density at the expense of some flowability and uniformity of the coalesced microstructure [54–55].

a Scanning electron micrograph (SEM) of virgin Duraform ProX PA powder. b Number size distribution estimated from SEM image (red bars) and Camsizer (gray bars), along with cumulative number distributions from SEM (red dotted line) and Camsizer (black solid line). c Volume size distributions from same data—legend applies to graphs b and c. d Number and volume distributions of particle sphericity from Camsizer; legend applies to graphs d and e. e Number and volume distributions of particle aspect ratio from Camsizer, defined as ratio of minimum chord diameter xc min to maximum Feret diameter xFe max, see definitions in text
Figure 4b plots the number distribution of particle sizes estimated by two different techniques: manual analysis of the SEM image in Fig. 4a (red bars) and Retsch Camsizer X2 (gray bars). Equivalent circle particle diameters dEC were estimated from the SEM image by taking the geometric mean of the particle height and width (measured along the x- and y-axes of the image) for 225 particles. This is equivalent to approximating each particle as an ellipse with long axis parallel to either the x- or y-axis of the image. This method tends to overpredict dEC for particles oriented at nonzero angles to the x- and y-axes of the image, as well as those with low convexity, such as star-like particles [56].
The Camsizer measurement is also an image-based technique, based on high-throughput analysis of over 2 × 106 separate particles imaged in 2D by a pair of high-resolution CCD cameras in a continuous fluidized gas jet. The projected area A of each particle is converted to equivalent circle diameter by the Camsizer software according to \( {d}_{EC}=\sqrt{4A/\pi } \), which is more accurate than the approximation described above used for SEM image analysis. In addition, the sample size for the Camsizer measurement is larger by a factor of nearly 104, and the Camsizer software automates the calculation of number and volume distributions (Fig. 4b, c) and shape parameters (Fig. 4d, e). Therefore, the Camsizer measurements are much more reliable than the estimates from SEM image analysis, but size distributions from both techniques are presented for comparison purposes.
Both SEM and Camsizer analyses result in multimodal number distributions nd as seen in Fig. 4b, with a small peak in each distribution centered around 50 μm and a larger peak between 1 and 10 μm. The Camsizer distribution features an even larger peak around 1 μm, whereas this small dEC peak is absent in the SEM number distribution. This peak may correspond to fumed silica, which is typically added to LS powders in the range of 0.1 wt% as a flow agent, or it may simply be a population of very fine polymer particles. The absence of this peak in the SEM image may be an artifact of the sample preparation, in which the powder sample was sprinkled onto adhesive tape followed by a short blast of nitrogen gas to remove weakly adhered particles, thus potentially removing the smallest fraction. Particle density is also much greater in the SEM image (Fig. 4a) than the images captured by the Camsizer, resulting in smaller particles tending to be covered up by larger ones. Nevertheless, the number distributions appear qualitatively similar above about 5 μm, as can be seen by the similar shape of the cumulative distributions in this range.
Volume distributions vd (Fig. 4c) were obtained from the number distributions in Fig. 4b using the center of each 2 μm bin to approximate dEC for the SEM analysis according to \( {v}_d=\frac{n_d{d}_{EC}^3}{\sum {n}_d{d}_{EC}^3} \). Due to the negligible contribution of the finest particle fraction, the volume distributions from both techniques appear virtually identical, other than some roughness on the high end of the SEM distribution due to the small sample size. The volume distribution is relatively narrow, featuring a d50 of about 60 μm with the majority of the distribution falling between 10 and 100 μm (d10 = 39 μm, d90 = 81 μm) as has been identified for optimal flowability in LS [57].
The Camsizer also calculates several shape parameters, including sphericity Sph, aspect ratio AR, and convexity C. These are defined as:
Here A is the projected area of each 2D particle image, P is the perimeter of A, and AC is the convex hull area [56]. Minimum chord diameter dc min is analogous to sieve diameter and is sometimes called the inner width of the particle projection. Maximum Feret diameter dFe max is the longest direct length measured between tangent lines, analogous to caliper measurement.
Figure 4d plots number and volume distributions of sphericity measured by the Camsizer, illustrating that the edges of Duraform particles are relatively smooth and round which facilitates good flowability for LS. Interestingly, the volume distribution is shifted to somewhat lower sphericity than number distribution, implying that larger particles tend to be less spherical than smaller ones. The number and volume distributions of aspect ratio, plotted in Fig. 4e, are more similar to one another and are centered around 0.6–0.8 indicating deviations from roundness consistent with the “potato”-like shape observed in Fig. 4a, in which many particles appear as a combination of two or more nominally spherical primary particles. Nearly all particles exhibit convexity > 0.95, supporting the relative accuracy of the elliptical approximation used for SEM analysis as discussed above.
Relative Molecular Weight
Measuring molecular weight of polyamides is challenging because they are insoluble in most common organic solvents. A common technique is to modify the amide groups with trifluoroacetic anhydride (Fig. 5), rendering the polymer soluble in solvents such as dimethylformamide (DMF) [44–45]. A size exclusion chromatography (SEC) trace of virgin Duraform thus modified is shown in Fig. 5, along with results of the relative number- and weight-averaged molecular weight (Mn and Mw, respectively) and dispersity (Đ, calculated as Mw/Mn) estimated from differential refractive index (dRI) calibrated relative to monodisperse polystyrene standards. The raw calibration results were converted to the equivalent polyamide molecular weight, reported in Fig. 5, by dividing by the molar weight ratio of the TFAA-modified monomer (293 Da) to the unmodified PA12 monomer (197 Da).

Size exclusion chromatography trace in dimethylformamide (DMF) of virgin Duraform ProX PA powder modified by reaction with trifluoroacetic anhydride (TFAA)
A wide range of molecular weight averages have been reported in the literature for virgin SLS grade PA12 determined by various SEC techniques, some of which are listed in Table 1. The absolute Mw reported recently by Wudy and Drummer [58] for PA2200 is considered most reliable because the unmodified polymer was dissolved directly in hexafluoroisopropanol (HFIP) without TFAA modification, and absolute molecular mass was determined by triple detection (light scattering, dRI, and an infrared concentration detector) via Mark-Houwink analysis, as opposed to more qualitative determination relative to PS standards reported here and by Zarringhalam et al. [55] It should be noted that the TFAA modification protocol used by Zarringhalam [55] and Dupin et al. [59] differs slightly from this work, which could result in a different degree of conversion for the reaction shown in Fig. 5. Hence, reported values from these reports should not be quantitatively compared even among papers using similar methods such as this one. Nevertheless, Zarringhalam’s study used the same protocol for both Duraform and PA2200, and the results suggest Duraform may have a slightly higher Mw. The relative Mw of approximately 10 kDa estimated in this work is less than 1/3 that of the absolute value reported by Wudy, and the Đ of 1.14 is significantly lower than previous reports. Future work is needed to determine whether this is an inaccuracy caused by poor solubility of TFAA-modified PA12 in DMF relative to polystyrene, or whether the ProX grade of Duraform has a lower molecular weight than earlier grades of Duraform, possibly to improve coalescence by means of a lower initial viscosity, as will be discussed in “Melt Rheology.”
Thermal and Hygroscopic Analysis
As a baseline comparison to the properties of LS-processed samples, virgin Duraform powder was also processed by compression molding on a programmable melt press (MP) at 230 °C, followed by controlled cooling at 5 °C/min and machining of test specimens from the resulting solid sheets. Thermogravimetric analysis (TGA, Fig. 6) of the virgin powder shows that degradation begins around 300 °C in nitrogen, well above the process temperatures used in both LS (169 °C) and MP (230 °C). The MP temperature was chosen as a likely upper limit for the temperature reached upon melting by the laser in LS. Although the MP process was performed in air, the polymer is expected to be stable in both processes from an oxidation perspective.

Thermogravimetric analysis (TGA) of virgin Duraform PA powder in nitrogen atmosphere, including a mass and b mass loss derivative. Temperatures of the LS powder bed setpoint and melt press are indicated by blue solid and red dashed vertical lines, respectively

Single cantilever mode dynamic mechanical analysis (DMA) of laser-sintered specimen 9 (Fig. 3) grip section milled to 1 mm thickness and 1-mm-thick melt-pressed samples either vacuum dried at 75 °C for 1.5 h or stored at 47% ± 11% relative humidity for 3 days before testing: a storage modulus E′, b loss modulus E″, and c loss tangent tan δ. Glass transition temperatures Tg are interpreted from the peak in tan δ
Dynamic mechanical analysis (DMA, Fig. 7) of LS and MP Duraform ProX samples reveals glassy state relaxations as well as the glass transition temperature Tg, interpreted here as the position of the largest peak in the bending mode loss tangent tan δ. Comparing a 1-mm-thick MP sample that was dried under vacuum at 75 °C for 1.5 h before testing to a specimen cut from the same MP sheet and stored at ambient conditions (47% ± 11% relative humidity, 21 °C ± 4 °C) for 3 days before testing, it can be seen that conditioning in the moderately humid lab environment causes only a 4 °C drop in Tg relative to the dried sample, which may be within the error of the measurement technique [61]. This is evidence of the relatively low moisture uptake of PA12, a significant advantage over more common polyamides such as PA66 whose Tg is known to drop by as much as 40 °C due to increasing relative humidity from 5 to just 40% [62]. Interestingly, the 1.3-mm-thick LS sample stored under the same ambient conditions exhibited Tg of 53 °C. While additional measurements are needed to establish the measurement error, this observation suggests PA12 parts produced by LS may be even less sensitive to moisture than conventionally processed PA12. This could be related to differences in crystallinity, as will be discussed in the next sections.
Differential scanning calorimetry (DSC) provides a wealth of information about LS material properties [63] and as a result is one of the most commonly reported characterization methods for LS PA12. Figure 8 shows DSC curves for first and second heat/cool cycles of virgin Duraform ProX PA, a sample cut from LS specimen 6 (Fig. 3), and a sample cut from the 1-mm-thick MP film. The initial temperature setpoint for the LS powder bed of 169 °C is represented by a vertical green line, illustrating that the powder is preheated to just below its melting onset as determined from the 10 °C/min DSC ramp. Thermal energy due to laser absorption pushes selected regions of the powder over this melt transition for a short time, yet when the powder returns to the bed temperature, it remains nearly 15 °C above its crystallization onset, again measured dynamically at 10 °C/min. Thus, melted regions of the layer recrystallize and solidify slowly [57, 64–65], enabling ample time for viscous coalescence and for mobile chain ends to interdiffuse both among particles within the current layer and downward into the recently melted layer below [66–67]. Slow recrystallization is also critical for avoiding uneven shrinkage which tends to result in distortion and curling of parts [57].

Differential scanning calorimetry (DSC) at 10 °C/min of virgin, laser-sintered (LS), and melt-pressed PA12: a first heat-cool cycle and b second heat-cool cycle. Green vertical line corresponds to LS bed temperature, and purple vertical line is reported Tm of 100% crystalline γ phase PA12 [33] (best viewed in color)
The so-called sintering window, the temperature difference between melt and crystallization peak onsets in DSC, is particularly wide for PA12 and is one of the reasons it is so successful in LS processing. However, the melting peak temperature of virgin Duraform is reduced by 7 °C on the second heating cycle (Fig. 8b), indicating that the melt temperature of the virgin powder has been intentionally increased to improve its processability for LS, such as by annealing to increase the crystalline lamellae thickness [38,51]. Furthermore the melt enthalpy of the virgin material (93 J/g) is more than double that observed on the second heat (40 J/g), meaning it is approximately 44% crystalline according to the heat of fusion of 210 J/g reported for 100% crystalline γ-phase PA12 [33]. In contrast, the MP sample exhibits a first heat melt temperature and enthalpy only slightly greater than the second heat values for virgin powder, owing to the use of a 5 °C/min cooling rate in the MP process rather than 10 °C/min in DSC, resulting in approximately 22% crystallinity compared with 19% for the second heat of all samples. These crystallinities are summarized in Table 2 and compared with measurements and predictions from other techniques which are discussed throughout the paper.
The LS specimen exhibits dual melting peaks, which are well known in the LS research community to represent a microstructure in which smaller particles and the outer shell of larger particles have melted and recrystallized, while the cores of larger particles remained unmelted by the laser [55, 59, 63]. This phenomenon tends to be more pronounced for Duraform powders due to their broader size distribution relative to powders such as PA2200, as discussed in “Particle Size and Shape Distributions.” The degree of particle melt (DPM) has been shown to increase with laser energy density and is correlated with increased ultimate tensile elongation [68]. DPM was estimated from
where the subscripts R and N refer to recrystallized and nascent (unmelted) material, respectively. Melt enthalpies ΔHR and ΔHN were obtained by fitting the two major melting peaks observed for LS specimen 6 with the function suggested by Dupin et al. [59] and integrating. The crystallinity XCN of the pure nascent phase was taken as 47% according to the average of DSC and X-ray diffraction (XRD) measurements for the virgin powder (Table 2), while crystallinity of fully melted and recrystallized Duraform in the LS process was assumed to be 30% [59]. The resulting DPM is approximately 86%, in good agreement with that found by Dupin et al. for Duraform at similar laser energy density De. This De represents the manufacturer’s suggested processing conditions, likely chosen to balance mechanical properties with fine feature resolution. While mechanical properties can be optimized by maximizing particle melting (higher De), above a certain De feature resolution is sacrificed due bleeding of thermal energy and molten material into the surrounding powder bed [43].
X-Ray Diffraction
The crystallinity of Duraform ProX was further investigated using XRD, shown in Fig. 9. The virgin powder exhibits a pair of primary peaks at diffraction angles 2Θ of 21.1° and 22.1° (Cu Kα source, λ = 1.54 Å), corresponding to crystal lattice d-spacings of approximately 4.02 Å and 4.21 Å according to Bragg’s law:

X-ray diffraction (XRD) spectra of the virgin Duraform ProX PA powder, 3.175-mm-thick melt-pressed section, and LS tensile specimen grip sections representing the four build orientations in Fig. 3
where n is an integer value. Interestingly, these d-spacings do not match those reported for any of the well-studied crystalline phases of PA12 such as the pseudohexagonal γ phase which typically exhibits a single peak around d = 4.15 Å [65, 69]. The spacings also do not match those expected for the monoclinic α phase (dual peaks at d = 3.6 Å and 4.5 Å) which has been obtained by crystallization from solution, crystallization from the melt under pressures > 500 MPa, or by drawing near the melt temperature [70,72,73,73], nor the α′ phase (dual peaks at d = 4.3 Å and 4.4 Å) which has only been observed near the melting point [65, 74–75]. Similar diffraction spectra corresponding to the 4.02 Å and 4.21 Å d-spacings have been reported for virgin PA2200 LS powder [76–77], which like Duraform is obtained by high-pressure precipitation from ethanol [51–52]. Although the authors of those studies referred to the structure variously as α and α′ [76–77], their d-spacings are distinct from those associated with the α form at equilibrium.
Dual XRD peaks around 4 Å in polyamides are typically associated with a mismatch between the interchain spacing (larger d) and the intersheet spacing (smaller d) (Fig. 2b, c). In α phase, the intersheet spacing is temperature-sensitive due to the significant mobility of the H-bonded sheets relative to one another, while the interchain spacing is relatively temperature-insensitive because it is fixed by the hydrogen bond length. In γ phase, these spacings are roughly equivalent because the H-bonds form across sheets at an angle of approximately 60°, leading to a single observed peak [32]. A pair of peaks with d = 4.0 Å and 4.2 Å have been reported for a wide range of polyamides at elevated temperatures approaching their Brill transition TBrill [32, 78], during which the structure transitions from α- to a γ-like pseudohexagonal arrangement via a hypothesized intermediate state in which the intersheet spacing systematically increases due to thermal expansion [32]. For imperfect crystals, d-spacings tend to be closer to each other than if approaching TBrill from the well-crystallized α form [32].
Therefore, we hypothesize that a relatively imperfect, nonequilibrium crystalline morphology that is intermediate between α and γ structures (such as is commonly observed during the Brill transition) forms and then becomes kinetically trapped as Duraform particles are precipitated from the rapidly stirred, pressurized ethanol solution used commercially to produce this powder between 100 and 125 °C [52]. These conditions are very different from those of Ishikawa and Nagai [71] for example, who observed well-resolved mixtures of α and γ phases for PA12 films formed by low-pressure evaporative casting from phenol/ethanol solution. Conversely, Monobe and Fujiwara [70] observed d-spacings of 4.0 Å and 4.1 Å (consistent with a quenched nonequilibrium Brill transition intermediate phase) upon precipitating PA12 from hot triethylene glycol, which is more comparable to the process used to make Duraform and PA2200. Interestingly, the PA12 LS powder sold by Farsoon appears to be much closer to the equilibrium α form, exhibiting well-resolved XRD peaks at 2Θ of 20° and 23.7° corresponding to d = 3.8 Å and 4.4 Å [79], significantly different than those seen here and for PA2200 [76–77].
Following melt pressing of Duraform ProX, a single peak is observed at 2Θ = 21.4°, characteristic of the γ form (d = 4.15 Å) expected for PA12 crystallized from the melt at low to moderate pressures. Interestingly, a similar peak is observed for the surface of LS specimen 6 (Y-X orientation, see caption of Fig. 3 for orientation scheme), whereas LS specimens 2 (Z-Y orientation), 12 (Y-Z orientation), and 27 (45° Z-Y orientation) all exhibit a combination of the γ peak superimposed with the 22.1° peak characteristic of the virgin powder intersheet spacing. Since the X-ray beam is diffracted from the broad horizontal surface of the specimens (Fig. 3), this result suggests the degree of particle melting is greater on the surface of a single LS layer (specimen 6) than at edges formed by many layer-layer interfaces (broad surface of all other specimens). As discussed in section S1 of the supplementary information, the X-ray penetration depth is on the order of a few hundred micrometers, meaning several LS layers contribute to the observed diffraction peak for specimen 6. Therefore, unmelted particle cores must contribute to the diffracted signal, since a degree of particle melting of 86% was estimated from the DSC results above, and such cores have been observed by several researchers to be periodically arrayed parallel to layer-layer welds with spacing of approximately 100 μm [59, 63, 80–81]. Figure S2 in the supplementary shows that a superposition of the virgin powder XRD spectra weighted by 21% along with the melt-pressed spectra weighted by 79% is qualitatively similar to the spectra of LS specimen 6. These proportions are consistent with a simple geometric model, described in the supplementary, in which unmelted particle cores reside at the bottom of each layer. This situation could arise due to attenuated laser penetration through the 100-μm-thick powder layers during LS processing, which was predicted by Drummer and coworkers [82]. On the other hand, the XRD spectra of specimen 2 are best fit by a superposition comprising 37% virgin powder and 63% melt-pressed Duraform, indicating a greater contribution from unmelted particles in specimens whose diffraction surface is formed by many welded layer edges (Fig. 3). This contribution likely arises from excess unmelted particles known to adhere at the outer edges of each melted layer slice during LS processing, additional evidence for which will be shown later in “Tensile Fractography.”
Crystallinity was estimated from the XRD spectra by fitting the region covering approximately 5° < 2Θ < 35° with a series of Lorentzian peaks, including an amorphous halo covering the entire range and either one or two narrow crystalline peaks depending on the observed morphology. The results, based on the ratio of integrated crystalline peaks to total peak area, are shown in Table 2 and agree relatively well with the crystallinity estimated from DSC.
Density
PA12 has the lowest density of commercially available nylons, with a typical datasheet value of 1.01–1.03 g/cm3 for injection-molded PA12. As measured using a helium pycnometer (results in Table 2), the MP Duraform ProX agrees closely with this value, while the virgin powder is significantly denser at 1.118 g/cm3 ± 0.003 g/cm3. Density ρ can be predicted from the measured crystallinity Xc according to
Here the subscripts a and c refer to densities of the pure amorphous and crystalline phases, respectively. Theoretical values of ρc for pure α and γ phase PA12 have been estimated from lattice parameters determined by X-ray crystallography [32–33], while ρa has been measured from quenched PA12 [33]. These literature values of ρc and ρa are listed in Table 2, along with predicted densities ρ for virgin, LS 6, and MP samples calculated from Eq. 7 using the average of Xc determined by DSC and XRD.
The value of ρc used in Eq. 7 depends on the relative content of α and γ phase. As discussed above, this is complicated by the fact that the virgin powders (and unmelted portions of the LS specimens) appear to be in an intermediate phase between α and γ whose density is unknown. For the purposes of estimation, we use an approximate ρc of 1.060 g/cm3 for this intermediate phase, based on the midpoint between α and γ values according to the simple rule of mixtures. For the LS specimen, the crystalline fraction is separated into intermediate and pure γ portions according to the DPM analysis discussed above, giving a final ρc of 1.039 g/cm3 in the LS material.
As shown in Table 2, densities predicted by this approach are systematically lower than those measured by pycnometry. It is tempting to assign these differences to error in the pycnometer measurement; however, it must be noted that the density predicted by Eq. 7 for the MP sample of 1.000 g/cm3 is lower than the range of 1.01–1.03 g/cm3 commonly cited for commercial PA12 (typically around 25% crystallinity [83]), whereas the pycnometer measurement is well within this range. The discrepancy is likely due to the two-phase model implicit in Eq. 7, which neglects partially ordered regions that are neither entirely crystalline nor amorphous. Furthermore, there are discrepancies in the values of ρc reported in the literature; for example, Ishikawa et al. [72] reported a ρc of 1.11 g/cm3 for pure γ PA12, which yields predicted values of ρ that are within about 1% of the measured values for MP and LS 6 specimens. As discussed in experimental “Pycnometry,” this is within the uncertainty of the pycnometer measurement.
The measured density of 1.118 g/cm3 for virgin powder exceeds prediction by over 8%, suggesting the powder may have absorbed a significant amount of moisture due to its high surface area. Dupin et al. [59] reported a density of 1.056 g/cm3 for Duraform powder measured by pycnometry. On the other hand, LS specimen 6 has a measured density of only 0.98 g/cm3, consistent with internal porosity which will be discussed in “Porosity Analysis by Computed Tomography.”
Vibrational Spectroscopy
Vibrational spectroscopy is a powerful and often used technique for identifying the chemistry and crystalline state of polyamides. Fourier transform Raman (FT-Raman) spectra of virgin Duraform ProX powder, an MP sample, and LS specimen 7 (Fig. 3), including both the rough printed surface and a milled surface cut into the interior of the LS sample, are shown in Fig. 10. ATR-FTIR spectra of the same samples are also shown in Fig. 3. ATR-FTIR is sensitive to vibrations of functional groups with strong dipoles such as N–H stretching (3250 cm−1), the C=O stretching amide I band (1630 cm−1), and the complex amide II band associated with coupling between –NH–C(O)– bending and C–N stretching (1500 to 1600 cm−1), as well as a number of CH2 vibrational modes [32]. FT-Raman on the other hand effectively screens out dipoles to emphasize CH2 deformations such as those observed at 1450 cm−1 and 1300 cm−1 which dominate the polarizability of the largely aliphatic PA12 chain’s electron cloud. From the dashed vertical lines in the figure, it can be seen that principal bands of the CO2 laser at 9.4 μm and 10.6 μm wavelength coincide with subtle fingerprint absorptions due to CH2 stretching and skeletal motions involving the CONH group [36, 71], resulting in a moderate amount of laser absorption during LS processing.

Fourier transform Raman spectra Fourier transform infrared (FTIR) attenuated total reflectance (ATR) spectra of the virgin Duraform ProX PA powder, 1-mm-thick melt-pressed section, and LS tensile specimen 7 grip section including the rough “sintered” surface as well as milled interior surface of the LS part
ATR-FTIR spectra reveal a number of subtle differences between the samples, the most obvious of which can be seen in the amide II bands (1500 to 1600 cm−1) which appear as a superposition of two major peaks with location and relative intensities that vary among samples. Approximate positions of these peaks as well as a number of other bands are listed in Table 3 where they are compared with expected positions for PA12 in both the α and γ crystalline forms according to a literature survey by Rhee and White [36]. Expanded views of the spectra showing these bands are in Figure S3 in the supplementary material. It should be noted that the uncertainty in peak positions cannot be estimated from the current analysis. While some spectral features appear to be superpositions of multiple peaks (Figure S3), detailed curve fitting to estimate error is outside the scope of this work. The following discussion is intended only to provide general observations and is not based on rigorously determined confidence intervals.
For virgin Duraform ProX powder, the majority of primary peaks are similar to those reported for γ phase PA12 (Table 3), including the lack of an amide V as this band is typically only observed in the α form [36]. On the other hand, the primary skeletal bend absorption at 541 cm−1 is close to that reported for α PA12, and the spectra exhibit a number of weaker secondary peaks characteristic of α phase including the amide I, amide III, and amide VI bands. A number of bands are closer to γ than α, but do not match the literature positions for either phase, including the hydrogen-bonded NH stretch (3292 cm−1), CH2 bend adjacent to the amide nitrogen (1459 cm−1), splitting of amide II (1195 cm−1), and CONH in plane vibration (948 cm−1). The virgin powder also exhibits a shoulder at 709 cm−1 near the CH2 rocking peak that is not seen in the MP sample and is not reported in the literature for any known phase of PA12. Since many of these shifts are thought to originate from the orientation of neighboring hydrogen-bonded chains (either parallel as in γ phase or antiparallel as in α) [36], the above observations tend to support the hypothesis developed in the XRD section that the virgin powder is in an intermediate semicrystalline phase between α and γ.
In contrast, the MP sample exhibits a number of characteristic α bands including amide I, amide II, amide V (there is a broad peak at 683 cm−1, unlike in the virgin spectra), and primary amide VI peak. However, the MP spectra also exhibit a roughly equal number of γ peaks (Table 3). MP exhibits only one peak shifted away from both phases (NH stretch) but at 3300 cm−1, it is closer to both γ and α than is the virgin powder. The mixture of α and γ peaks is confusing since XRD indicates MP is primarily γ phase. Rhee and White [36] observed a similar situation for unannealed biaxially oriented PA12 films which exhibited XRD spectra indicative of γ but a number of α characteristic FTIR bands. They hypothesized that the α bands in FTIR arise due to amorphous chains hydrogen bonded in an antiparallel arrangement, similar to the α crystalline phase. These bands disappeared upon annealing in Rhee and White’s study, while the XRD spectra did not change. It is worth noting that the MP material in this study is crystallized under a moderate amount of pressure (10 MPa) which may be sufficient to facilitate a degree of antiparallel organization in the amorphous phase as a precursor to α crystallization. The semicrystalline α phase has been reported to form when melt crystallized above 500 MPa pressure [70,72,73,73].
The LS specimens exhibit a mix of these two situations, such as the amide II peaks which exhibit features similar to both virgin and MP spectra. The surface spectrum of LS specimen 7 is more similar to that of the virgin powder, while the milled side is more similar to the MP specimen, as seen in the positions of the amide I, N vicinity CH2 bending, splitting of amide II, CONH in plane, and skeletal bend bands (although the surface appears to feature a weak amide V band similar to that of the MP spectra). These observations are generally consistent with the presence of unmelted particles adhering to the surface of LS parts as well as a smaller fraction of unmelted particle cores in the sample interior. The ATR penetration depth is only 1 to 2 μm, a small fraction of the particle d50 of 50 μm and much lower than XRD which samples several 100 μm powder layers as discussed above.
There are no significant peak position differences found in the Raman spectra. Nevertheless, Hendra and coworkers [84] identified the ratio of the FT-Raman CH2 bending peak intensity around 1440 cm−1 to the amide I band around 1640 cm−1 as a convenient indicator of the number of CH2 groups per monomer in n-polyamides. They proposed that the peak at 1440 cm−1 corresponds to CH2 groups in the middle of the aliphatic chain, enabling straightforward determination of the chain length for unknown nylons. According to their method, PA12 is expected to have a ratio I1440/I1640 equal to about 5. For all Duraform ProX samples studied here, this ratio was 5.0 ± 0.3, in good agreement with Fig. 10 of their paper [84].
Melt Rheology
Coalescence in LS depends critically on zero shear viscosity η0, since little pressure or shear is applied to consolidate the molten powder [60, 85]. A typical design target is for η0 to remain between 103 and 104 Pa s for up to 10 min following melting by the laser [86]. Parallel plate oscillatory rheology was used in an attempt to measure η0 of molten Duraform ProX material. Temperature was maintained at 200 °C, which approximates the temperature reached following melting by the laser in LS [87]. As shown in Fig. 11a, complex viscosity η* failed to reach a plateau during a positive frequency sweep from 10−1 rad/s to 102 rad/s, so it was followed by a negative frequency sweep back to 10−1 rad/s. Within the time taken to complete these two steps (26 min), low-frequency viscosity increased by a factor of 4, consistent with the well-known chemical aging of LS grades of PA12 in which unprotected amine and carboxylic acid chain end groups react via condensation to increase the polymer molecular mass at a rate that increases with temperature (Fig. 2a) [38, 58, 76]. As has been discussed in the literature, this scheme enables PA12 to coalesce well in LS due to an initially low η0 while improving ultimate mechanical properties by increasing molecular weight and interlayer bonding following coalescence. However, chemical aging also limits powder recyclability since the reaction occurs in the solid state as well, increasing η0 of the used powder and necessitating continual blending with virgin powder to avoid degrading fine detail resolution, mechanical and surface properties, and repeatability of part performance [76]. Further evidence of chemical aging is shown in Fig. 11b, in which storage modulus G′ and loss modulus G″ rise continually during steady oscillation at 200 °C and 10 rad/s, until the curves cross after approximately 2.5 h. Similar results have recently been observed by Craft et al. [88] for PA2200.

Parallel plate oscillatory melt rheology of virgin Duraform ProX PA powder at 200 °C. a Positive and negative frequency sweeps. b Time sweep at constant angular frequency ω of 10 rad/s
Porosity Analysis by Computed Tomography
It is well known that LS PA12 parts tend to be porous, with typical void fractions on the order of 5 vol% [42, 59, 81, 89,91,91]. Porosity is thought to result from a number of factors, including insufficient packing of the powder bed, poor flow and coalescence due to high η0, trapped atmospheric gas (N2) between particles and layers, offgassing of degradation products from PA12 due to laser overexposure, and offgassing of water from PA12, either due to insufficient drying of the powder or from the product of condensation reactions among chain ends. Figure 12 shows X-ray micro-computed tomography (X-μCT) slices of four LS specimens representing the four build orientations in Fig. 3. All specimens exhibit a high density of voids on the order of the particle size. Visually, voids tend to be anisotropic and oriented parallel to layer-layer interfaces, as can be particularly seen in Fig. 12b, c in which the layer-layer interfaces run from left to right and top to bottom, respectively. This motivates a full 3D analysis of pore orientation.

X-ray micro-computed tomography (X μ-CT) slices of untested gauge cross sections of LS tensile specimens representing the four build orientations in Fig. 3: aZ-Y specimen 3, bY-X specimen 8, cY-Z specimen 13, and d 45° Z-Y specimen 28
For all four samples, Table 4 lists the total porosity, the number of pores analyzed for each sample, and the volume-based average values of L, W, T, and their ratios (defined in experimental “X-Ray Micro-computed Tomography”). In Table 4, the uncertainty in the porosities, due mainly to uncertainty in the chosen threshold, is about 0.7% (i.e., for specimen 3, the porosity is 7.4% ± 0.7%). The uncertainty in the L, W, and T parameters is about 2 voxels or 15 μm, arising from segmentation and the algorithm for determining these parameters. Note that all the values in Table 4 for specimens 8 (Y-X) and 13 (Y-Z) are similar to each other, and all the values for specimens 3 (Z-Y) and 28 (45° Z-Y) are similar to each other, yet both pairs of values are different from each other. The average aspect ratios show that these pores are not by any means spherical, so that their orientation is important to assess.
Figure 13 shows both the pore number-based and pore volume-based size distributions, where size in this case is chosen to be the volume-equivalent-spherical diameter (VESD). Other choices for the particle size, e.g., L, W, or T, would mainly shift the graphs horizontally, with much smaller changes in the vertical coordinate [92]. For random-shaped pores, no one dimension can fully capture the pore size, so different dimensions are more or less useful for different applications. The VESD for a given pore is defined as the diameter of the sphere that has volume equal to the pore’s volume. The volume-based graph is more clear than the number-based graph—all four pore size distributions are quite similar in shape, but the 8 and 13 pair, which were built with their tensile axes parallel to layer-layer interfaces (normal to Z), are centered at a smaller VESD than the 3 and 28 pair, which were built normal to the layer-layer interfaces. On average, the pores for the 8 and 13 pair are about 14% smaller than for the 3 and 28 pair. The average VESD is approximately given by the average of <L>, <W>, and <T>, which is about 158 μm for the 3-28 pair and about 136 μm for the 8-13 pair, in agreement with Fig. 13.

a Number-based pore size distribution and b volume-based pore size distribution for all four samples, with size defined as the volume-equivalent-spherical diameter (VESD, μm)
The difference in average pore size may be related to the geometry of the slices rastered by the laser during LS processing of each layer. For the horizontally oriented specimens 8 and 13, each layer’s slice comprises a 115-mm-long 2D projection of the dogbone shape oriented along the Y-axis, whereas for the vertically oriented specimens 3 and 28, the slices are much smaller rectangles corresponding to the 3 mm × 6 mm dogbone cross-section for specimen 3, or a 4.24 mm × 6 mm rectangle in the case of the 45° tilted specimen 28. The larger average pore size for these smaller scan areas may be associated with incomplete laser coverage due to turning at the edges of the scan area, or the interface between the outline and interior hatching scan regions, which is a substantially greater proportion of the total scan area for specimens 3 and 28.
Computation of pore orientation was done in three dimensions using the L, W, and T triad, which are measured along three mutually perpendicular directions and can thus serve as a measure of each void’s orientation. The unit vectors specifying L, W, and T were computed for each pore. For each of the four samples, distribution histograms were constructed for each component (ux, uy, uz) for L, W, and T. We use the unit vectors for L to define the orientation of a void, since L is the longest dimension. In the X-ray CT, the samples were all oriented in the same way so the unit vectors for all four samples originally shared the same coordinate system. However, based on Fig. 3, the samples were all oriented differently with respect to the defined coordinate axes in the LS build chamber, so the coordinates of the unit vectors for all the pores in the four samples were adjusted to match the coordinate system in Fig. 3.
After the L unit vector (ux, uy, uz) was computed for a pore, the sign of the Z-component was evaluated. If it was positive, it was not changed. If it was negative, the signs of all the components of the unit vector were inverted. This was done since these pores are 3D bodies. Orientation can be indicated by looking at either end. The Y-components were then evaluated. If the Y-component was positive, no change was made. If the Y-component was negative, then the signs of both the X- and Y-components were inverted. This assures that the projection of the full 3D unit vector in the X-Y plane maintains the orientation of the object. All this means that the limits of uz and uy are (0,1), while the limits for ux are (− 1,1).
Figure 14 shows the volume-based distribution histograms for all three components of the L unit vector, for all four samples but grouped into the specimen pairs 3-28 (ZY and 45° ZY) and 8-13 (YX and YZ). It is clear that the orientations of the members of these pairs are similar to each other but somewhat different from the other pair. In Fig. 3, Z is the common build direction (normal to layer-layer interfaces). Looking at the uz histograms, it is clear that the pores in all four samples tend to be oriented perpendicular to the build direction (parallel to layer-layer interfaces), since all four histograms are strongly peaked near uz = 0, though somewhat less so for the 8-13 pair and especially specimen 13 which exhibits a weak secondary peak near uz = 1. This effect could be related to the scan strategy in which laser raster path was alternated between X- and Y-directions every other layer. The scan area for specimen 13 is the most asymmetric of the four geometries, featuring a long Y-dimension of 115 mm compared to just 3 mm in the X-direction. The interior hatching region is even smaller considering the 0.5 mm outline scan on either edge of each slice, resulting in an interior X scan length of just 2 mm, or four laser line widths. Therefore, the extreme mismatch in scan length from layer to layer may have resulted in a small population of vertically oriented pores for this part orientation.

Volume-based distribution histograms for the components of the L unit vectors for all the identified pores, grouped into the specimen pairs 3-28 (ZY and 45° ZY, a–c) and 8-13 (YX and YZ, d–f)
The ux distributions for both pairs are almost perfectly symmetrical around zero, which can be seen visually and also since the volume-weighted average of ux is 0.0016 (specimen 3), 0.00023 (specimen 8), − 0.0046 (specimen 13), and 0.039 (specimen 28). There are peaks for the ux distributions at − 1 and 1, indicating a tendency for the longest part of the pore to lie along the X-axis (Fig. 3), although there is significant weight at every value of ux. This may occur because the long axes of particles (Fig. 4) become oriented along the X-axis (traversing direction of the rolling powder applicator), tending to result in pores with preferential orientation along this same direction.
The uy distributions show the most difference between the two sample pairs, with the specimens 3 and 28 fairly strongly oriented parallel to the Y-direction, while the 8-13 pair distribution is almost flat, indicating lack of preferential pore orientation with respect to the Y-axis. This difference may arise because of the much smaller scan area of the vertically oriented specimens 3 and 28 as discussed above. Dewulf et al. [91] observed a ring of increased porosity separating the fully dense outer contour scan region of a layer from the hatched interior scans of parts made from PA2200 on an EOS LS machine. While such a ring is not immediately obvious in Fig. 12a, d, as noted above, this interfacial region is a much greater proportion of the total scan area for specimens 3 and 28 than for the other specimen pair. Since slices for these specimens are oriented with their longest dimension along the Y-axis, the interface between outline and interior scans may result in preferential alignment of pores along the Y-axis for these orientations.
A final question about orientation is this: are the pores flattened, so that the thinnest direction tends to be in the build or Z-direction? Figure 14 just says that the longest part of the pore tends to be oriented perpendicular to the Z-direction (parallel to layer-layer interfaces). However, W, the next longest pore dimension, while perpendicular to L, could be oriented in the Z-direction, meaning the thinnest dimension T would not be. Figure 15 shows the volume-based distribution histogram for uz for W and T. It is clear that W, like L, is primarily perpendicular to the Z-direction, since the histogram is strongly peaked near uz = 0, while T is aligned with the Z-direction, since its uz histogram is very strongly peaked near uz = 1. Therefore, the thinnest part of each asymmetric pore indeed tends to be in the build direction, suggesting the pores tend to be oriented as flattened “pancakes” lying parallel to the LS layer planes.

Volume-based distribution histogram for uz for W (a) and T (b) for all four LS samples
Orientation of pores parallel to LS layer-layer interfaces has been noted by a few authors [59, 81], although to our knowledge the degree of pore orientation has not been quantified in previous studies. Note that the cross-sectional view of specimen 3 (ZY) in Fig. 12a is oriented parallel to LS layer-layer interfaces, causing the voids to appear randomly oriented, while the cross section of specimen 28 in Fig. 12d is at a 45° angle to layer interfaces and hence a moderate degree of orientation is observed. Periodically coplanar porosity, with voids organized in planes parallel to layer-layer interfaces, has been found for a number of different PA12 powders processed on a variety of machines, [59, 81, 91] and this is also observed in Fig. 12b, c to some extent. Earlier studies [80] supposed coplanar porosity to arise from poor layer-layer welding, but it is now recognized that the Z-oriented tensile strength of LS parts is typically higher than would be expected in that case. More recently, coplanar porosity was hypothesized to arise from entrapped gas that rises like soda bubbles in the molten polymer regions before becoming trapped by unmelted particle cores [81], which typically are coplanar along layer-layer interfaces [51, 54, 68–69]. The “pancake”-like pore shape suggested by Fig. 15 may occur due to buoyancy forces according to the “soda bubble” hypothesis discussed above, or it may be a consequence of compressive force applied by the roller during fresh layer applications.
Tensile Mechanical Properties
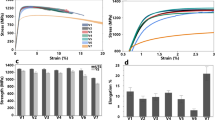
Tensile properties are an important determinant of part performance and are one of the primary test methods used by manufacturers to certify LS builds [1]. Figure 16a shows tensile stress-strain curves measured using digital image correlation for melt-pressed Duraform ProX and representative LS specimens from each of the four orientations shown in Fig. 3. Young’s modulus E (Fig. 16b), ultimate tensile strength σUlt (Fig. 16c), and ultimate elongation εUlt (Fig. 16d) were calculated from these curves; the reported figures represent averages and standard deviations of four LS specimens from each orientation and four MP specimens. Melt-pressed Duraform ProX is clearly stronger and more ductile than the same material when processed by LS, although it is substantially less ductile than injection-molded (IM) PA12. Rüsenberg et al. [89] reported IM PA2200 with elongation of 250%, but ultimate strength of only 42.5 MPa, considerably lower than the strength of MP Duraform ProX. These differences are likely related to the long slow cooling step of 40 min at 5 °C/min used in the MP process in this work, facilitating the growth of large spherulites and high crystallinity (relative to IM PA12), both of which tend to reduce ductility in semicrystalline polymers. In contrast, the IM process used by Rüsenberg involved a cooling time of just 25 s at a mold temperature of 50 °C, likely resulting in primarily amorphous PA12 which would be expected to have higher ductility and lower strength.

Tensile mechanical properties of Duraform ProX PA specimens: a stress-strain curves of melt-pressed and laser-sintered specimens representing each of the four build orientations of Fig. 3. Average b Young’s modulus, c ultimate tensile strength, and d ultimate elongation of four specimens from each category (six for melt pressed), where error bars represent ± one standard deviation
The LS PA12 tensile parameters are very much in line with those reported in the literature [47, 81, 91, 93–94]. Z-oriented (vertical) tensile elongation is typically half or less that of parts printed parallel to layer-layer interfaces. This is thought to result from a combination of factors including weaker molecular welding between layers, subcritical crack propagation at the edges of anisotropic coplanar pores oriented parallel to layer planes (perpendicular to stress for vertical specimens), and reduced ductility of unmolten particle cores which also tend to be coplanar along layer-layer interfaces. It should be noted that mechanical properties can be optimized by using a higher laser energy density that fully melts all particle cores, eliminating several of these limitations and achieving Z elongation in excess of 20% [81]. However, in practice, this is usually not done because it results in very poor dimensional accuracy and detail resolution due to bleeding of thermal energy and material flow into the surrounding powder [43].
Tensile Fractography
Fracture surfaces of the tensile specimens featured in Fig. 16a were imaged using scanning electron microscopy (SEM), revealing a mix of ductile and brittle fracture regions as shown in Fig. 17. The MP fracture surface is primarily ductile, featuring large regions of highly stretched and fibrillated material consistent with the high elongation of this sample (top row of Fig. 17a–c). LS fracture surfaces on the other hand appear primarily brittle indicative of catastrophic fast fracture, but each LS specimen features a small edge region containing fibrillated and stretched material indicative of a more ductile failure mode. These regions, circled in the low magnification full surface images in the left column of Fig. 17, are likely the sites of fracture initiation during the tensile test. Higher magnification images in the middle and right columns reveal information about deformation mechanisms of the unique microstructures resulting from LS processing. Vertical (ZY) specimen 5 has layer-layer interfaces parallel to the failure plane, exhibiting wide shallow pores (Fig. 17e) whose interior surfaces feature round nodes that may be unmelted particle cores (Fig. 17f, detail of e). Stretched material at the pore edges in Fig. 17e may be evidence of subcritical crack growth leading ultimately to catastrophic failure, which as discussed above is a likely factor in the low elongation of vertically oriented LS samples.

a–o Scanning electron microscope (SEM) images of tensile fracture surfaces. Left column features stitched low-resolution images representing the entire fracture surface of each specimen indicated to the left of each row. Middle and right columns show high-resolution details of regions on these surfaces
YX (horizontal, Fig. 17h) specimen 7 features horizontal rows of < 50 μm nodes with a unique fibrillated structure that may be fractured unmolten particle cores arranged parallel to layer-layer interfaces. These densely fibrillated nodes are separated by regular rows of coarser melted and recrystallized polymer which is also stretched. Specimen 15 (YZ, horizontal edge orientation, Fig. 17k) appears similar but with rows oriented vertically, again parallel to layer-layer interfaces. In both samples, pores are visible pinned against the rows of unmolten cores and in some cases straddling rows, consistent with the “soda bubble” hypothesis discussed above. Brittle fracture regions reveal a more random mix of what appear to be unmolten cores (Fig. 17i) and γ phase melted and recrystallized spherulites (Fig. 17l), indicative of fast fracture in these regions that does not follow the layered microstructure. The 45° vertical specimen 29 has a large ductile region featuring a mix of morphologies similar to fracture patterns of horizontal (Fig. 17n, compare to h) and vertically oriented (Fig. 17o, compare to e) samples.
Figure 18 features side views of the same failed specimens, revealing elongational effects of tensile deformation whose direction is indicated by arrows in Fig. 18. The upper left-hand image shows that the gauge width of the MP specimen has necked near the failure point, likely coinciding with the strain softening observed around 15% strain in Fig. 16a. The MP side surface is mostly featureless, but a few regions of moderate fibrillation are observed as shown in the top row of Fig. 18. No strain softening occurred for any LS specimens (Fig. 16a), and correspondingly there is little evidence of necking for them in the left column of Fig. 18. Higher magnification images in the middle column show the printed surface morphology of LS specimens, revealing layers of unmelted or partially melted particles adhering to the dense part surfaces, a well-known drawback of LS. Interestingly, the surface of horizontal (YX) specimen 7 appears considerably more melted and smooth; this surface is formed by a single powder layer whereas the other three LS surfaces shown consist of multiple layer-layer interfaces at the edge of scan areas where unwanted powder adhesion occurs. The surface of specimen 7 features numerous craze-like cracks that propagate through the centers of partially melted particles rather than around their boundaries, providing evidence that powder welding is very strong within a single layer. These craze-like features are largely absent from the cross-layer surfaces of specimens 5 and 15, consistent with poorer interlayer bonding. The 45° ZY specimen 29 is in between these two behaviors, exhibiting smaller craze-like fractures separating just one or two particles from the dense surface, likely due to the stair-stepping effect in which periodic regions of the surface are formed by single layers followed by more widely spaced layer interfaces.

Scanning electron microscope (SEM) images of front (broad) surfaces of fractured tensile specimens. Left column features stitched low-resolution images representing the full width of each specimen indicated to the left of each row (arrows indicate tensile direction). Middle and right columns show high-resolution details of regions on these surfaces
Conclusions
We have presented a wide range of physical property measurements for PA12 powder and specimens formed both by conventional melt processing and by laser sintering on a modern production LS machine that has not been widely studied in the scientific PA12 LS literature. Results are discussed in the context of this literature, agreeing with many previous observations while offering new insights such as the size and shape distributions of powder particles, localization of unmelted particle cores as identified by the XRD signature of apparently nonequilibrium crystalline phase in the virgin powder, time dependence of PA12 melt viscosity as a consequence of chemical aging, quantified anisotropy and orientation of pores in LS specimens, and fractographic observations of unique deformation mechanisms depending on the orientation of stress relative to processing directions. These measurements will serve as the foundation of a public data set intended to aid modelers and practitioners in mitigating risks and reducing costs of manufacturing with LS through use of predictive simulations and data-based build strategies.
Notes
Identification of specific commercial equipment or materials in this publication is not intended to imply recommendation or endorsement by the Army Research Laboratory, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose
Certain commercial equipment, instruments, or materials are identified in this paper in order to specify the experimental procedure adequately. Such identification is not intended to imply recommendation or endorsement by the National Institute of Standards and Technology, nor is it intended to imply that the materials or equipment identified are necessarily the best available for the purpose.
References
(2014) ASTM F3091-14 standard specification for powder bed fusion of plastic materials. ASTM International: West Conshohocken
Tofail SAM, Koumoulos EP, Bandyopadhyay A, Bose S, O’Donoghue L, Charitidis C (2018) Additive manufacturing: scientific and technological challenges, market uptake and opportunities. Mater Today 21(1):22–37
Huang Y, Leu MC, Mazumder J, Donmez A (2015) Additive manufacturing: current state, future potential, gaps and needs, and recommendations. J Manuf Sci Eng 137(1):014001–014001-10
Gao W, Zhang Y, Ramanujan D, Ramani K, Chen Y, Williams CB, Wang CCL, Shin YC, Zhang S, Zavattieri PD (2015) The status, challenges, and future of additive manufacturing in engineering. Computer Aided Design 69:65–89
Hopkinson N (2006) Production economics of rapid manufacture. In: Hopkinson N, Hague RJM, Dickens P (eds) Rapid manufacturing: an industrial revolution for the digital age. Wiley, West Sussex, pp 147–158
Goodridge RD, Tuck CJ, Hague RJM (2012) Laser sintering of polyamides and other polymers. Prog Mater Sci 57(2):229–267
Yuan S, Shen F, Chua CK, Zhou K (2018) Polymeric composites for powder-based additive manufacturing: materials and applications. Prog Polym Sci 91:141–168
Schmid M (2018) Laser sintering with plastics. Carl Hanser Verlag GmbH & Co. KG, Munich
Fox B (2006) Rapid manufacture in the aeronautical industry. In: Hopkinson N, Hague RJM, Dickens P (eds) Rapid manufacturing: an industrial revolution for the digital age. Wiley, West Sussex, pp 221–232
Wooten J (2006) Aeronautical case studies using rapid manufacture. In: Hopkinson N, Hague RJM, Dickens P (eds) Rapid manufacturing: an industrial revolution for the digital age. Wiley, West Sussex, pp 233–240
Lyons B (2012) Additive manufacturing in aerospace: examples and research outlook. The Bridge: Linking Engineering and Society 42(1):13–19
Khajavi SH, Partanen J, Holmström J (2014) Additive manufacturing in the spare parts supply chain. Comput Ind 65(1):50–63
Spielman R (2006) Space applications. In: Hopkinson N, Hague RJM, Dickens P (eds) Rapid manufacturing: an industrial revolution for the digital age. Wiley, West Sussex, pp 241–248
Reuters, world’s biggest truck maker turns to 3D printing for spare parts. Fortune 2016, http://fortune.com/2016/07/13/daimler-trucks-3d-printing-spare/. Accessed 31 Oct 2018
LS allows 928 motorsports to improve Porsche 928 performance. Stratasys. https://www.stratasysdirect.com/resources/case-studies/3d-printing-selective-laser-sintering-porsche-engine-928-motorsports. Accessed 31 Oct 2018
Jin Y-a, Plott J, Chen R, Wensman J, Shih A (2015) Additive manufacturing of custom orthoses and prostheses—a review. Procedia CIRP 36:199–204
Deckers JP, Vermandel M, Geldhof J, Vasiliauskaite E, Forward M, Plasschaert F (2017) Development and clinical evaluation of laser-sintered ankle foot orthoses. Plast, Rubber Compos 47(1):42–46
Faustini MC, Neptune RR, Crawford RH, Stanhope SJ (2008) Manufacture of passive dynamic ankle-foot orthoses using selective laser sintering. IEEE Trans Biomed Eng 55(2 Pt 1):784–790
Harper NG, Russell EM, Wilken JM, Neptune RR (2014) Selective laser sintered versus carbon fiber passive-dynamic ankle-foot orthoses: a comparison of patient walking performance. J Biomech Eng 136(9):091001–091001–7
South BJ, Fey NP, Bosker G, Neptune RR (2009) Manufacture of energy storage and return prosthetic feet using selective laser sintering. J Biomech Eng 132(1):015001–015001-6
Idaho steel embraces 3D printing to deliver superior-quality parts faster. 3D Systems. https://www.3dsystems.com/learning-center/case-studies/idaho-steel-embraces-3d-printing-deliver-superior-quality-parts-faster. Accessed 31 Oct 2018
Balzereit S, Proes F, Altstädt V, Emmelmann C (2018) Properties of copper modified polyamide 12-powders and their potential for the use as laser direct structurable electronic circuit carriers. Addit. Manuf. 23:347–354
Beta LAYOUT—multi-dimensional circuit carriers using additive manufacturing. EOS. https://www.eos.info/case_studies/beta-layout-3d-printed-multidemsional-circuit-carriers. Accessed 31 Oct 2018
New Balance pushes the limits of innovation with 3D printing (2013) New Balance. https://www.newbalance.com/press-releases/id/press_2013_New_Balance_Pushes_Limits_of_Innovation_with_3D_Printing.html. Accessed 31 Oct 2018
Nike debuts first-ever football cleat built using 3D printing technology (2013) Nike News. https://news.nike.com/news/nike-debuts-first-ever-football-cleat-built-using-3d-printing-technology. Accessed 31 Oct 2018
Nike football accelerates innovation with 3D printed “Concept Cleat” for Shuttle (2014) Nike News. https://news.nike.com/news/nike-football-accelerates-innovation-with-3d-printed-concept-cleat-for-shuttle. Accessed 31 Oct 2018
The future of running is here (2016) New Balance. https://www.newbalance.com/article?id=4041. Accessed 31 Oct 2018
Kinematics dress (2014) Nervous System. https://n-e-r-v-o-u-s.com/projects/sets/kinematics-dress/. Accessed 31 Oct 2018
SLS design guide. 3D Systems. https://www.3dsystems.com/sites/default/files/2017-01/3dsystems-sls-designguide-2016.pdf. Accessed 31 Oct 2018
Bourell DL, Watt TJ, Leigh DK, Fulcher B (2014) Performance limitations in polymer laser sintering. Phys Procedia 56:147–156
Niino T, Sato K (2009) Effect of powder compaction in plastic laser sintering fabrication. Proceedings of the Solid Freeform Fabrication Symposium, pp 193–205
Aharoni SM (1997) n-Nylons: their synthesis, structure, and properties. Wiley, Chichester
Puffr R, Raab M, Dolezel B (1991) Properties. In: Puffr R, Kubanek V (eds) Lactam-based polyamides volume I: polymerization, structure, and properties. CRC Press, Boca Raton, pp 187–260
Holmes DR, Bunn CW, Smith DJ (1955) The crystal structure of polycaproamide: nylon 6. J Polym Sci 17(84):159–177
Bradbury EM, Brown L, Elliott A, Parry DAD (1965) The structure of the gamma form of polycaproamide (nylon 6). Polymer 6(9):465–482
Rhee S, White JL (2002) Crystal structure and morphology of biaxially oriented polyamide 12 films. J Polym Sci B Polym Phys 40(12):1189–1200
Inoue K, Hoshino S (1973) Crystal structure of nylon 12. J Polym Sci Polym Phys Ed 11(6):1077–1089
Schmid M, Amado A, Wegener K (2014) Materials perspective of polymers for additive manufacturing with selective laser sintering. J Mater Res 29(17):1824–1832
Kruth JP, Vandenbroucke B, Van Vaerenbergh J, Mercelis P (2005) Benchmarking of different SLS/SLM processes as rapid manufacturing techniques. Int Conf Polymers & Moulds Innovations
Seepersad CC, Govett T, Kim K, Lundin M, Pinero D (2012) A designer’s guide for dimensioning and tolerancing SLS parts. Proceedings of the Solid Freeform Fabrication Symposium, pp 921–931
Akande SO, Dalgarno KW, Munguia J, Pallari J (2016) Assessment of tests for use in process and quality control systems for selective laser sintering of polyamide powders. J Mater Process Technol 229:549–561
Pavan M, Craeghs T, Verhelst R, Ducatteeuw O, Kruth J-P, Dewulf W (2016) CT-based quality control of laser sintering of polymers. Case Studies in Nondestructive Testing and Evaluation 6:62–68
Schmid M, Kleijnen R, Vetterli M, Wegener K (2017) Influence of the origin of polyamide 12 powder on the laser sintering process and laser sintered parts. Appl Sci 7(5):462
Jacobi E, Schuttenberg H, Schulz RC (1980) A new method for gel permeation chromatography of polyamides. Makromol. Chem., Rapid Commun 1(6):397–402
Cureton LT, Napadensky E, Annunziato C, La Scala JJ (2017) The effect of furan molecular units on the glass transition and thermal degradation temperatures of polyamides. J Appl Polym Sci 134(46):45514
ISO/ASTM 52921-13 (2013) Standard terminology for additive manufacturing-coordinate systems and test methodologies. ASTM International, West Conshohocken
Usher JS, Gornet TJ, Starr TL (2013) Weibull growth modeling of laser-sintered nylon 12. Rapid Prototyp J 19(4):300–306
Tamari S (2004) Optimum design of the constant-volume gas pycnometer for determining the volume of solid particles. Meas Sci Technol 15(3):549–558
Taylor MA, Garboczi EJ, Erdogan ST, Fowler DW (2006) Some properties of irregular 3-D particles. Powder Technol 162(1):1–15
Van den Eynde M, Verbelen L, Van Puyvelde P (2015) Assessing polymer powder flow for the application of laser sintering. Powder Technol 286:151–155
Scholten H, Christoph W (1997) Use of a nylon-12 for selective laser sintering. US6245281B1, Evonik Degussa GmbH, EOS GmbH Electro Optical Systems
Meyer K-R, Hornung K-H, Feldmann R, Smigerski H-J (1979) Method for polytropically precipitating polyamide powder coating compositions where the polyamides have at least 10 aliphatically bound carbon atoms per carbonamide group. US4334056A, Huels AG
Baumann F-E, Monsheimer S, Grebe M, Christoph W, Heinrich D, Renners H, Scholten H, Schiffer T, Muegge J, Chiovaro J (2002) Polyamide powder with long-lasting, consistently good flowability. US7211615B2, Evonik Degussa GmbH
Dickens ED, Lee B-L, Taylor GA, Magistro AJ, Ng H, McAlea KP, Forderhase PF (1992) Sinterable semi-crystalline powder and near-fully dense article formed therewith. USRE39354E1, 3D Systems Inc.
Zarringhalam H, Hopkinson N, Kamperman NF, de Vlieger JJ (2006) Effects of processing on microstructure and properties of SLS nylon 12. Mater Sci Eng A 435-436:172–180
Garboczi EJ, Bullard JW (2013) Contact function, uniform-thickness shell volume, and convexity measure for 3D star-shaped random particles. Powder Technol 237:191–201
Amado A (2016) Characterization and prediction of SLS processability of polymer powders with respect to powder flow and part warpage. Ph.D. Thesis, ETH Zürich, Zürich, Switzerland
Wudy K, Drummer D (2019) Aging effects of polyamide 12 in selective laser sintering: molecular weight distribution and thermal properties. Additive Manufacturing 25:1–9
Dupin S, Lame O, Barrès C, Charmeau J-Y (2012) Microstructural origin of physical and mechanical properties of polyamide 12 processed by laser sintering. Eur Polym J 48(9):1611–1621
Haworth B, Hopkinson N, Hitt D, Zhong X (2013) Shear viscosity measurements on polyamide-12 polymers for laser sintering. Rapid Prototyp J 19(1):28–36
McAninch IM, Palmese GR, Lenhart JL, La Scala JJ (2015) DMA testing of epoxy resins: the importance of dimensions. Polym Eng Sci 55(12):2761–2774
Launay A, Marco Y, Maitournam MH, Raoult I (2013) Modelling the influence of temperature and relative humidity on the time-dependent mechanical behaviour of a short glass fibre reinforced polyamide. Mech Mater 56:1–10
Zarringhalam H, Majewski C, Hopkinson N (2009) Degree of particle melt in nylon-12 selective laser-sintered parts. Rapid Prototyp J 15(2):126–132
Zhao M, Wudy K, Drummer D (2018) Crystallization kinetics of polyamide 12 during selective laser sintering. Polymers 10(2):168
Paolucci F, Baeten D, Roozemond PC, Goderis B, Peters GWM (2018) Quantification of isothermal crystallization of polyamide 12: modelling of crystallization kinetics and phase composition. Polymer 155:187–198
Kline DB, Wool RP (1988) Polymer welding relations investigated by a lap shear joint method. Polym Eng Sci 28(1):52–57
Seppala JE, Hoon Han S, Hillgartner KE, Davis CS, Migler KB (2017) Weld formation during material extrusion additive manufacturing. Soft Matter 13(38):6761–6769
Majewski C, Zarringhalam H, Hopkinson N (2008) Effect of the degree of particle melt on mechanical properties in selective laser-sintered nylon-12 parts. Proc Inst Mech Eng B J Eng Manuf 222:1055–1064
Dencheva N, Nunes TG, Oliveira MJ, Denchev Z (2005) Crystalline structure of polyamide 12 as revealed by solid-state13C NMR and synchrotron WAXS and SAXS. J Polym Sci B Polym Phys 43(24):3720–3733
Monobe K, Fujiwara Y (1967) A study on single crystals of nylon 12. Memoirs of the School of Engineering, Okayama University, 2(1):88–92
Ishikawa T, Nagai S, Kasai N (1980) Effect of casting conditions on polymorphism of nylon-12. J Polym Sci Polym Phys Ed 18(2):291–299
Ishikawa T, Nagai S, Kasai N (1981) The γ → α partial transformation in nylon 12 by drawing. Die Makromolekulare Chemie 182(3):977–988
Nobuyasu H, Kazunao H, Susumu H (1983) Study of transformations among α, γ and γ' forms in nylon 12 by X-ray and DSC. Jpn J Appl Phys 22(2R):335
Li LB, Koch MHJ, de Jeu WH (2003) Crystalline structure and morphology in nylon-12: a small- and wide-angle X-ray scattering study. Macromolecules 36(5):1626–1632
Ramesh C (1999) Crystalline transitions in nylon 12. Macromolecules 32(17):5704–5706
Dadbakhsh S, Verbelen L, Verkinderen O, Strobbe D, Van Puyvelde P, Kruth J-P (2017) Effect of PA12 powder reuse on coalescence behaviour and microstructure of SLS parts. Eur Polym J 92:250–262
Xu F, Yan C, Shyng Y-T, Chang H, Xia Y, Zhu Y (2014) Ultra-toughened nylon 12 nanocomposites reinforced with IF-WS 2. Nanotechnology 25(32):325701
Jones NA, Atkins EDT, Hill MJ, Cooper SJ, Franco L (1996) Chain-folded lamellar crystals of aliphatic polyamides. Comparisons between nylons 4 4, 6 4, 8 4, 10 4, and 12 4. Macromolecules 29(18):6011–6018
Wu J, Xu X, Zhao Z, Wang M, Zhang J (2018) Study in performance and morphology of polyamide 12 produced by selective laser sintering technology. Rapid Prototyp J 24(5):813–820
Leigh DK (2012) A comparison of polyamide 11 mechanical properties between laser sintering and traditional molding. Proceedings of the Solid Freeform Fabrication Symposium:574–605
Stichel T, Frick T, Laumer T, Tenner F, Hausotte T, Merklein M, Schmidt M (2017) A round robin study for selective laser sintering of polyamide 12: microstructural origin of the mechanical properties. Opt Laser Technol 89:31–40
Osmanlic F, Wudy K, Laumer T, Schmidt M, Drummer D, Körner C (2018) Modeling of laser beam absorption in a polymer powder bed. Polymers 10(7):784
Murray BR, Leen SB, Semprimoschnig COA, Brádaigh CMÓ (2016) Helium permeability of polymer materials as liners for composite overwrapped pressure vessels. J Appl Polym Sci 133(29)
Hendra PJ, Maddams WF, Royaud IAM, Willis HA, Zichy V (1990) The application of Fourier transform Raman spectroscopy to the identification and characterization of polyamides—I. Single number nylons. Spectrochim Acta A: Mol Spectrosc 46(5):747–756
Verbelen L, Dadbakhsh S, Van den Eynde M, Kruth J-P, Goderis B, Van Puyvelde P (2016) Characterization of polyamide powders for determination of laser sintering processability. Eur Polym J 75:163–174
Brule B, Decraemer N (2014) Method for producing an object by melting a polymer powder in a powder sintering device. US20180001549A1, Arkema France
Greiner S, Wudy K, Wörz A, Drummer D (2019) Thermographic investigation of laser-induced temperature fields in selective laser beam melting of polymers. Opt Laser Technol 109:569–576
Craft G, Nussbaum J, Crane N, Harmon JP (2018) Impact of extended sintering times on mechanical properties in PA-12 parts produced by powderbed fusion processes. Addit Manuf 22:800–806
Rüsenberg S, Schmidt L, Schmid H-J (2011) Mechanical and physical properties—a way to assess quality of laser sintered parts. Proceedings of the Solid Freeform Fabrication Symposium, pp 239–251
Rouholamin D, Hopkinson N (2014) An investigation on the suitability of micro-computed tomography as a non-destructive technique to assess the morphology of laser sintered nylon 12 parts. Proc Inst Mech Eng B J Eng Manuf 228(12):1529–1542
Dewulf W, Pavan M, Craeghs T, Kruth J-P (2016) Using X-ray computed tomography to improve the porosity level of polyamide-12 laser sintered parts. CIRP Ann 65(1):205–208
Erdoğan ST, Garboczi EJ, Fowler DW (2007) Shape and size of microfine aggregates: X-ray microcomputed tomography vs. laser diffraction. Powder Technol 177(2):53–63
Majewski C, Hopkinson N (2011) Effect of section thickness and build orientation on tensile properties and material characteristics of laser sintered nylon-12 parts. Rapid Prototyp J 17(3):176–180
Starr TL, Gornet TJ, Usher JS (2011) The effect of process conditions on mechanical properties of laser-sintered nylon. Rapid Prototyp J 17(6):418–423
Acknowledgments
The authors wish to thank Matt Bartucci, Daniel Cole, John La Scala, and Jian Yu for useful discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Official contribution of the National Institute of Standards and Technology; no subject to copyright in the United States
Electronic Supplementary Material
ESM 1
(DOCX 710 kb)
Rights and permissions
About this article

Cite this article
Bain, E.D., Garboczi, E.J., Seppala, J.E. et al. AMB2018-04: Benchmark Physical Property Measurements for Powder Bed Fusion Additive Manufacturing of Polyamide 12. Integr Mater Manuf Innov 8, 335–361 (2019). https://doi.org/10.1007/s40192-019-00146-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40192-019-00146-3