
Abstract
Purpose of Review
The Ras/mitogen-activated protein kinase (MAPK) pathway is essential in the regulation of cell cycle, differentiation, growth, cell senescence and apoptosis, all of which are critical to normal development. A class of neurodevelopmental disorders, RASopathies, is caused by germline mutations in genes of the Ras/MAPK pathway. Through the use of whole exome sequencing and targeted sequencing of selected genes in cohorts of panel-negative RASopathy patients, several new genes have been identified.
Recent Findings
New genes have been identified and include RIT1, SOS2, RASA2, RRAS and SYNGAP1, that likely represent new, albeit rare, causative RASopathy genes. In addition, A2ML1, LZTR1, MYST4, SPRY1 and MAP3K8 may represent new rare genes for RASopathies, but, additional functional studies regarding the mutations are warranted. In addition, recent reports have demonstrated that chromosomal copy number variation in regions encompassing Ras/MAPK pathway genes may be a novel pathogenetic mechanism expanding the RASopathies.
Summary
The identification of potential new genes and chromosomal copy number variation being associated with the RASopathies is very exciting and broadens our understanding of the biology of Ras signaling and the RASopathies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The RASopathies
The RASopathies are a class of developmental disorders that are caused by germline mutations in genes which encode components of the Ras/mitogen-activated protein kinase (MAPK) pathway [1•]. This includes the core pathway components and both positive and negative regulators of Ras signaling. The Ras/MAPK pathway is a ubiquitous, highly conserved, intracellular signaling pathway that is critical in cell cycle regulation, differentiation, growth, apoptosis, and cell senescence. It plays an essential role throughout mammalian development, from embryogenesis to tissue-specific cellular homeostasis in the adult. Therefore, it is not surprising germline mutations that affect its function can have a profound deleterious effect on development. Taken together, the RASopathies represent one of the most prevalent groups of congenital malformation syndromes affecting approximately 1 in 1000 individuals. These syndromes include (1) neurofibromatosis type 1 (NF1), caused by loss of function mutations in the protein neurofibromin [2–4]; (2) Noonan syndrome (NS) caused by mutations in PTPN11 [5], SOS1 [6, 7], RAF1 [8, 9], KRAS [10], NRAS [11], SHOC2 [12], and CBL [13, 14]; (3) NS with multiple lentigines caused by mutations in PTPN11 [15] and RAF1 [9]; (4) Legius syndrome caused by inactivating mutations in the SPRED1 gene [16]; (5) Costello syndrome (CS) caused by activating mutations in HRAS [17]; (6) cardio-facio-cutaneous syndrome (CFC) caused by gain of function mutations in BRAF [18, 19] and MAP2K1 or MAP2K2 [19]; and (7) capillary malformation-arteriovenous malformation (CM-AVM) caused by loss of function mutations in the RASA1 gene [20]. The RASopathies share many overlapping characteristics including craniofacial dysmorphology; cardiac, cutaneous, skeletal, and ocular abnormalities; neurocognitive impairment; hypotonia and impaired growth; and, in many, an increased cancer risk, because they all share the common pathogenetic mechanism of Ras/MAPK pathway signaling dysregulation and/or activation. However, because each of the syndromes is due to mutations in specific affected genes of the pathway, and distinct mutations within that gene, each has unique phenotypic features that result from the specific fashion by which the Ras/MAPK pathway is dysregulated.
Ras Signaling
The Ras proteins are small guanosine nucleotide-bound GTPases that act as a central signaling hub for multiple intracellular signaling pathways. Ras genes exist as a multigene family that includes HRAS, NRAS, and KRAS. Ras proteins cycle between an active guanosine triphosphate (GTP)-bound form and an inactive guanosine diphosphate (GDP)-bound form. Ras signals through two well-characterized effector pathways, the Ras/MAPK (Raf–MEK–ERK) and the phosphatidylinositol-3-kinase (PI3K) pathway, as well as through numerous other effector pathways including RalGDS, TIAM, PLCε, and AF6 [21]. Functional studies of mutant proteins associated with the RASopathies indicate that the common pathogenetic mechanism is activation and dysregulation of the Ras/MAPK pathway [1•]. Activation of Ras begins with the binding of growth factors to receptor tyrosine kinases (RTKs), G-protein coupled receptors, or other receptors that result in the recruitment of guanine-nucleotide exchange factor (GEF) son of sevenless (SOS) to the membrane. At rest, SOS is complexed with the adapter protein GRB2 (growth factor receptor-bound protein 2). Recruitment is mediated by the SH2 domain of GBR2 which interacts with the activated receptor tyrosine kinase (RTK). This brings SOS into the vicinity of Ras leading to the exchange of GDP for GTP, thereby switching Ras into the active state. Although Ras proteins have an intrinsic GTPase activity, it is too low to effectively inactivate Ras. Therefore, the conversion to the inactive state requires proteins that accelerate GTP hydrolysis, and GTPase-activating proteins (GAPs) such as neurofibromin or RASA1. Activated Ras leads to the activation of Raf proteins. Raf phosphorylates and activates MEK1/MEK2 (MAPK kinase) which then phosphorylates and activates the terminal MAPKs of the pathway ERK1 and/or ERK2. ERK1/2 function on a large number of downstream molecules, both nuclear and cytosolic. ERK1/2 substrates include transcription factors, membrane proteins, and protein kinases that, in turn, control essential cellular functions [22].
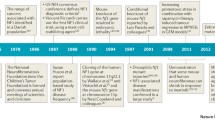
Although activation of the Ras/MAPK pathway has been shown to be the predominant biochemical hallmark of the RASopathies, aberrant Ras signaling through other effector pathways is likely to also play a significant role [23, 24]. In addition, the evolution of our understanding of the complexity of Ras signaling, and the increased availability of new powerful tools of discovery, such as exome/genome sequencing, will inevitably lead to the expansion of the pathogenic etiology of the RASopathies. Numerous patients have phenotypic features suggestive of a RASopathy; however, they have no mutations in any of the known RASopathy causative genes. For example, the pathogenetic cause of approximately 20 % of patients with the clinical diagnosis of RASopathy or a RASopathy-like clinical phenotype remains unknown. In this review, we highlight recent studies that have identified a number of new genes and potential new genes associated with the RASopathies (Table 1).
New Genes Associated with RASopathies
RIT1
Aoki et al. examined a NS cohort of 180 individuals with no known NS mutations [25]. They identified 17 individuals with nine unique missense mutations in RIT1 by whole exome sequencing and targeted sequencing of RIT1. Ras-like protein in tissues (RIT1) is a member of a novel branch of Ras-related GTPase proteins in the Ras family. Other members in the Ras subfamily include Ras-like protein in neurons (RIN) and Ras-like protein that interacts with calmodulin (RIC) [26]. All share approximately 50 % structural homology with Ras but lack a C-terminal lipidation site. The nine missense mutations include c.104G > C encoding p.S35T; c.176C > G encoding p.A57G; c.242A > G encoding p.Q81G; c.244T > G encoding p.F82 V; c.246T > G encoding p.F82L; c.247A > C encoding p.T83P; c.265T > C encoding p.Y89H; c.270G > T encoding p.M90I; and c.284G > C encoding p.G95A. Many of these RIT1 mutants and two additional missense mutants (p.A77G and p.A77P) were identified in follow-up studies in other NS cohorts with no known NS causing gene mutations [27, 28, 29•]. Most of the RIT1 mutations are in the switch I or II regions and are predicted to result in a constitutively active Ras protein. RIT1 has been shown to activate the Ras/MAPK pathway [30]. Functional analysis of RIT1 mutations by transient transfection into NIH3T3 and HEK293 cells demonstrated increased signaling of the Ras/MAPK pathway [25, 29•].
SOS2
Yamamoto et al. in 2015 identified two de novo heterozygous missense-causing mutations in SOS2 by whole exome sequencing in a cohort of 58 probands with the clinical diagnosis of NS, but who were negative for known NS mutations [31]. SOS2 encodes the protein son of sevenless 2, a RasGEF and homologue to SOS1, the second most frequently mutated gene in NS. The SOS2 mutations identified, p.M267K and p.T376S, are both located in the Dbl homology (DH) domain responsible for maintaining SOS2 in an autoinhibited conformation. In a subsequent study, targeted sequencing of SOS2 was carried out in an additional cohort of 150 clinically diagnosed NS patients also negative for known NS mutations [32]. They confirmed the SOS2 p.T376S missense mutation in four individuals, one of which was maternally inherited. In addition, they identified two novel de novo missense mutations: p.M267R and p.T264K. The p.M267 location in SOS2 is homologous to the p.M269 location in SOS1, a common mutation site in NS [31]. SOS2 mutants p.T264K, p.M267R, and p.T376S were functionally assessed in vitro using HEK293 cells. All three mutants resulted in higher levels of GTP-bound Ras and increased signaling of the Ras/MAPK pathway consistent with the known mechanism of the NS causative SOS1 mutations [32].
RASA2
In order to identify additional genes that may be associated with NS, Chen et al. [29•] performed whole exome sequencing on a cohort of 27 patients with a NS clinical diagnosis that did not have any mutations in known NS causing genes. They identified three patients with novel missense-causing mutations in the RASA2 gene. RASA2 encodes the RasGAP protein Ras P21 protein activator 2, RASA2. The three mutations identified affected two different amino acid residues p.Y326 and p.R511 of which both are in the conserved GAP domain (p.Y326C, p.Y326N, and p.R511C). The p.R511C substitution is in the Ras interaction site and is predicted to act as a dominant-negative competitor for Ras binding. Therefore, the mutations are assumed to result in loss of function. Overexpression of RASA2 mutant proteins in HEK293 cells resulted in prolonged Ras/MAPK signaling following epidermal growth factor stimulation. Loss of function mutations in RasGAPs are responsible for other RASopathies including NF1 caused by mutations in NF1 [2, 3, 33] and CM-AVM caused by mutations in RASA1 which encodes p120 RasGAP1 [20]. Moreover, missense mutations in RASA2 have been identified in human melanomas in addition to other cancers [34].
RRAS
Flex et al. in 2014 [35] sought to identify additional RASopathy-associated genes by screening for mutations in a selected group of candidate genes based on their predicted protein interaction/function with known RASopathy-associated proteins. In a RASopathy cohort of 96 unrelated individuals with no known RASopathy-associated mutations, they identified one missense-causing mutation in the RRAS gene in a patient described as having some phenotypic features of NS. The mutation, c.163G > A, results in a p.V55M residue substitution. It was not established if this mutation was de novo since the parents were not examined. This mutation was reported to occur in 1/13,000 alleles by the NHLBI exome sequencing project. The RRAS gene encodes RRAS (related ras viral (R-Ras) oncogene homologue) that exhibits 50–60 % homology to the Ras proteins. It is associated with several diverse cellular processes, including neuronal axon guidance, angiogenesis, cell adhesion, and migration. Activating mutations in RRAS are associated with numerous cancers [36]. Sequencing of RRAS in an additional cohort of 408 patients with a RASopathy phenotype who were also negative for known RASopathy gene mutations revealed one individual with a heterozygous 3 nucleotide duplication, c.116_118dup resulting in p.G39dup. Functional analysis of the mutant RRAS protein showed a decrease in intrinsic GTPase activity in an in vitro assay. Overexpression of mutant proteins in COS-7 cells increased Ras/MAPK pathway signaling, consistent with other RASopathy causing gene mutations. Therefore, activating mutations in RRAS may represent a rare RASopathy-associated gene with a NS phenotype.
SYNGAP1—A New RASopathy
RasGAPs (GTPase-activating proteins) are negative regulators of the Ras/MAPK pathway. Haploinsufficiency in neurofibromin (encoded by the NF1 gene), p120 RasGAP (encoded by the RASA1 gene), and Ras P21 protein activator 2 (encoded by the RASA2 gene) have all been found to cause RASopathies. SynGAP1 (synaptic Ras GTPase-activating protein 1) is a recently described RasGAP thought to be expressed only in neurons (for recent review see [37•]). Germline mutations in SYNGAP1 have been identified to cause autosomal dominant intellectual disability type 5 which is considered a nonsyndromic form of intellectual disability [38]. De novo heterozygous frame shift mutations were identified in individuals with nondysmorphic features, global delay, and hypotonia who also had moderate to severe intellectual disability. Other phenotypic features that were identified in the original cohort include seizures and ophthalmologic findings. More recently, expansion of the phenotype came to include global developmental delay with behavioral issues (aggressive behavior, sleep disturbances, and hyperexcitability) and autism spectrum disorder [39]. Mild dysmorphic features were also described and included microcephaly, myopathic facies, deep set eyes, long nose, and long ears with protuberant lobes. Additional features may include pectus, kyphosis, hip dysplasia, strabismus, fine hirsutism, and significant constipation with failure to thrive.
SYNGAP1, encoded by SYNGAP1, is located on chromosome 6p21.3. It is a major component of the postsynaptic density found associated with excitatory N-methyl-d-aspartate (NMDA) receptors at synapses. Its GAP domain is homologous to that of p120 RasGap and neurofibromin, two RasGAPs known to be associated with RASopathies. SYNGAP1 is phosphorylated by calmodulin-dependent protein kinase II, CaMKII, causing a decrease in its RasGAP activity resulting in an activation of the Ras/MAPK pathway. Mouse models of SynGAP1 +/− mice demonstrate alteration in Ras/MAPK signaling, synaptic plasticity, and learning, and activated ERK in hippocampal extracts [40]. The behavioral phenotype of SynGAP1 +/− mice, which express roughly half the normal levels of SynGAP1, exhibits multiple behavioral traits such as aberrant cognitive and noncognitive processes that are normally mediated by the hippocampus. Mice are also hyperactive, have reduced spatial alternation, and have severe working and reference memory deficits [41].
Although patients with SYNGAP1 mutations have several phenotypic features characteristic of upregulation of the Ras/MAPK pathway, individuals with SYNGAP mutations have not been noted to have the classical craniofacial phenotypic features which traditionally have been associated with RASopathies. Based on the unique neurologically centered clinical phenotype, the identification of causative mutations in a new RasGAP gene with neuronal restricted expression, and the fact that it does not categorize with any established RASopathy, we consider this a new RASopathy.
Candidate RASopathy Genes
A2ML1
Using whole exome sequencing in both the proband and parents to detect de novo variants, Vissers et al. reported a mutation in the A2ML1 gene in a proband with a clinical diagnosis of NS. The mutation, c.2405G > A, resulted in a p.R802H substitution [42]. A2ML1 which encodes the protein alpha-2-macroglobulin-like 1 (A2ML1) is a secreted broad-range protease inhibitor [43]. Autoantibodies to A2ML1 have been reported to be responsible for paraneoplastic pemphigus (PNP), a multi-organ syndrome characterized by intractable stomatitis, polymorphous cutaneous lesions, and lympho-proliferative tumors [44]. However, the c.2405G > A variant was also reported in 4/12,194 alleles in the NHLBI exome sequencing project. This initial variant finding was expanded upon by targeted sequencing of A2ML1 in a cohort of 155 NS patients with no identified RASopathy causing mutations. They identified two individuals with missense mutations in A2ML1: One with the same mutation they previously found, c.2405G > A encoding a p.R802H missense substitution, and a second mutation c.1775G > T encoding p.R592L. Both of these were shown to segregate with the individuals offspring who also exhibited a NS-like phenotype. The individual with the p.R802H mutation had inherited it from his mother, but surprisingly she was reported to exhibit no RASopathy phenotype. Both the mutations are predicted to impair protein function. In addition, a study examining the genotype–phenotype correlation for external ear anomalies and hearing impairment in a NS cohort identified one individual with a c.4061 + 1G > A mutation in A2ML1 [45]. The functional consequences of the A2ML1 mutations on Ras/MAPK pathway signaling were examined by overexpression transfection experiments in HEK293T and COS7 cells. However, neither mutation resulted in increased phosphorylated ERK levels [42]. The authors theorized that one binding target of A2ML1 is lipoprotein receptor-related protein 1 (LRP1) which is an upstream activator of the Ras/MAPK pathway. LRP1 associates with SHC domain proteins and CBL during recruitment to the plasma membrane [46, 47]. These findings suggest that rare A2ML1 mutations may be associated with a RASopathy exhibiting a highly variable NS-like phenotype; however, further studies are needed to confirm the causality.
LZTR1
Yamamoto and colleagues identified five missense-causing variants in LZTR1 by whole exome sequencing in a cohort of 58 probands with the clinical diagnosis of NS, but who were negative for known NS mutations [31]. The missense mutations consisted of p.G248R p.R284C, p.R287Y, p.Y119C, and p.S247N. Of these, two of the mutants p.G248R and p.S247N were also shown to segregate with the NS phenotype within two large families. In addition, Chen and colleagues identified two heterozygous LZTR1 missense mutations in similar regions (p.R237Q and p.A249P) by exome sequencing in their NS patient cohort [29•]. However, the functional characterization of these missense mutations was not commented upon. The LZTR1 (leucine-zipper-like transcriptional regulator 1) protein belongs to a functionally diverse family of proteins containing BTB-kelch domains that is thought to localize to the cytoplasmic surface of the Golgi membrane [48]. All of the mutations are in the highly conserved kelch domain and are predicted to disrupt protein function. Kelch domains form a tertiary structure that may also be responsible for binding to other proteins [49]. Somatic LZTR1 mutations have been described in glioblastoma. In addition, germline LZTR1 mutations are associated with schwannomatosis, one of the neurofibromatoses characterized by late onset tumor predisposition [50]. These finding suggest that mutations in LZTR1 may be responsible for a rare percentage of NS cases. However, it is not known if the identified LZTR1 mutations increase Ras/MAPK signaling, the underlying pathogenetic mechanism of the RASopathies.
MYST4
Karyotyping of a patient with a clinical diagnosis of NS who did not have any identified mutations in genes known to be causative of NS revealed a balanced de novo chromosomal translocation t(10;13) (q22.3;q34) [51]. The translocation breakpoint was mapped to 10q22.3 to a location disrupting the MYST4 gene causing MYST4 haploinsufficiency. The MYST4 gene, also known as KAT6B, encodes MYST histone acetyltransferase (monocytic leukemia) 4 (also termed, K(Lysine) acetyltransferase 6B). Histone acetyltransferases modify DNA by transferring an acetyl group from acetyl-CoA to histone proteins on DNA. This epigenetic modification of DNA plays an important role in gene regulation. Generally, increased histone acetylation globally results in increased transcriptional activation. The authors demonstrated in a lymphoblastoid cell line derived from the patient that haploinsufficiency of MYST4 resulted in an increase of Ras/MAPK pathway activity. In addition, genome-wide chromosome immunoprecipitation analysis demonstrated that gene targets of MYST4 were somewhat biased toward MAPK signaling pathways. The authors postulated that altered expression of multiple genes associated with Ras/MAPK pathway regulation may be responsible for the increase in pathway activation and the NS-like phenotype. However, more research is necessary to confirm this novel correlation.
SPRY1
A nonsense de novo mutation, p.E79*, in SPRY1 was identified by whole exome sequencing in one individual in a cohort of 27 clinically diagnosed NS patients with no known NS associated mutations [29•]. SPRY1 encodes the protein Sprouty1 which is a negative regulator of Ras/MAPK pathway signaling. The complete mechanism by which Sprouty1 inhibits Ras/MAPK signaling remains unclear, it is to thought to act at the level of the signaling from the RTK to Ras [29•]. Functional studies need to be carried out to establish whether or not this SPRY1 nonsense mutation results in increased Ras/MAPK pathway signaling.
MAP3K8
Chen et al. identified a de novo missense mutation p.L128V in MAP3K8 in one individual in the 27 NS patient cohort described above. MAP3K8 encodes mitogen-activated protein kinase kinase kinase 8 (MAP3K8) which has been shown to cause lung cancer and can directly activate MEK1 [52]. The significance of the p.L128V mutation is not known because although it is in a conserved region, the function of the region is not known. However, functional analysis by overexpression of the mutant protein in HEK293 cells resulted in increased levels of phosphorylated ERK. Additional studies in additional RASopathy cohorts will need to be carried out to confirm the pathogenetic significance of this gene.
Copy Number Variation Associated with RASopathy Genes
Over the past several years, rare copy number variants of genes encoding Ras pathway components have been reported in individuals with phenotypic features reminiscent of RASopathy syndromes. Multiple studies have identified chromosomal duplications encompassing PTPN11 in patients with a clinical diagnosis of NS [53–55]. PTPN11 encodes SHP2 which facilitates Ras activation. Gain of function missense mutations in SHP2 are the most common mutations associated with NS, accounting for approximately 50 % of all cases. Chromosomal duplication of region 12q24.13 encompassing the PTPN11 gene was detected by array comparative genomic hybridization (array CGH) [53, 54]. Moreover, a de novo trisomy of 12q24.11q24.21 encompassing the PTPN11 gene was recently reported [55]. In each of these cases, gene duplication is thought to result in increased dosage of SHP2 which has the equivalent effect as the common gain of function mutation in PTPN11 associated with NS. In addition, rare chromosomal duplications associated with SHOC2 in a NS patient [56], and RAF1 and MAP2K2 in patients with mild RASopathy spectrum phenotypes have been reported [57, 58]. Chromosomal deletions have also been reported to be associated with a RASopathy phenotype. A chromosomal deletion encompassing the BRAF locus at 7q34 was reported in two individuals with phenotypes overlapping several RASopathies [58, 59]. In addition, two studies reported interstitial deletion of 19p13.3 encompassing the MEK2 gene [60, 61]. The patients had clinical features suggestive of dysregulation of the Ras/MAPK pathway but were not classical for CFC or any of the other RASopathies. Functional studies of the MEK2 deletion caused MEK2 haploinsufficiency and resulted in dysregulation of the Ras/MAPK pathway [60].
Conclusion
The initial identification of the genes causative for RASopathies which exhibit overlapping phenotypic features, also exhibit many disparate characteristics as well, has enabled us to classify these syndromes as to their common underlying pathogenetic etiology, the dysregulation of the Ras/MAPK pathway. The RASopathies caused by germline mutations in genes encoding components of the Ras/MAPK pathway underscore the essential role the pathway plays in normal embryonic and postnatal development, since dysregulation of the pathway has severe developmental consequences. In addition, the number of different affected genes and the diversity of mutations within each gene are reflected in the variety of affected phenotypic features present in this class of syndromes. The accurate identification of a causative gene mutation for a given genetic disorder is essential in establishing a molecular diagnosis corresponding to the clinical diagnosis and enables the physician to accurately manage the health of the patient. Numerous patients have phenotypic features suggestive of a RASopathy; however, they have no mutations in any of the known RASopathy causative genes. Recent advances in genome sequencing technology provide unprecedented capabilities for the discovery of rare genes causative for genetic disorders. In particular, the modest cost of whole exome sequencing has made it increasingly useful in this endeavor. With the application of this technology, the identification of a number of genes potentially causative for RASopathies has expanded. Several of the recently identified genes associated with the RASopathies including, RIT1, SOS2, RASA2, RRAS and SYNGAP1, likely represent new, albeit rare, causative RASopathy genes. They were identified in multiple patients, some segregated with the phenotype, and the function of their encoded protein clearly links them to Ras signaling. In addition, several genes were identified that are potentially associated with the RASopathies; however, their link to Ras signaling is novel, and their link to Ras signaling is not entirely understood. These include A2ML1, LZTR1, and MYST4. They may represent new rare genes causative for RASopathies, but additional studies regarding the function of their encoded proteins and the effects of the mutations are warranted. The identification of these genes being associated with the RASopathies is very exciting and broadens our understanding of the biology of Ras signaling and the RASopathies. Because of the increased application of genome and exome sequencing, based on next generation sequencing technology to discover new genetic causes for genetic disorders, it is challenging to determine if the detected gene variant is truly pathogenetic. Therefore, the College of Medical Genetics and Genomics and the Association for Molecular Pathology have recently addressed this challenge and have suggested standards and guidelines for interpretation of the sequence variants [62•]. Utilizing this framework will help standardize the criteria for the assignment of a new gene variant as being causative for a given disorder. In addition, since the underlying biochemical phenotype associated with the RASopathies is activation of the Ras/MAPK pathway, functional studies regarding new gene variants are essential in establishing their causality.
References
Papers of particular interest, published recently are highlighted as: • Of importance
• Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–69. This is a current review of the RASopathies including phenotypic features and genetics.
Wallace MR, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249(4965):181–6.
Cawthon RM, et al. Identification and characterization of transcripts from the neurofibromatosis 1 region: the sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics. 1990;7(4):555–65.
Viskochil D, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62(1):187–92.
Tartaglia M, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–8.
Roberts AE, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39(1):70–4.
Tartaglia M, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39(1):75–9.
Razzaque MA, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39(8):1013–7.
Pandit B, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007–12.
Schubbert S, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38(3):331–6.
Cirstea IC, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42(1):27–9.
Cordeddu V, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41(9):1022–6.
Niemeyer CM, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42(9):794–800.
Martinelli S, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet. 2010;87(2):250–7.
Digilio MC, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71(2):389–94.
Brems H, et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39(9):1120–6.
Aoki Y, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37(10):1038–40.
Niihori T, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38(3):294–6.
Rodriguez-Viciana P, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311(5765):1287–90.
Eerola I, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73(6):1240–9.
Rajalingam K, et al. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773(8):1177–95.
Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24(1):21–44.
Marin TM, et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest. 2011;121(3):1026–43.
Chen PC, et al. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome-associated Sos1 mutation. J Clin Invest. 2010;120(12):4353–65.
Aoki Y, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173–80.
Rusyn EV, et al. Rit, a non-lipid-modified Ras-related protein, transforms NIH3T3 cells without activating the ERK, JNK, p38 MAPK or PI3K/Akt pathways. Oncogene. 2000;19(41):4685–94.
Bertola DR, et al. Further evidence of the importance of RIT1 in Noonan syndrome. Am J Med Genet A. 2014;164A(11):2952–7.
Gos M, et al. Contribution of RIT1 mutations to the pathogenesis of Noonan syndrome: four new cases and further evidence of heterogeneity. Am J Med Genet A. 2014;164A(9):2310–6.
• Chen PC, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci USA. 2014;111(31):11473–8. This is a very comprehensive study using whole-exome-sequencing and gene targeted sequencing to discover new genes associated with the RASopathies.
Shi GX, Andres DA. Rit contributes to nerve growth factor-induced neuronal differentiation via activation of B-Raf-extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades. Mol Cell Biol. 2005;25(2):830–46.
Yamamoto GL, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015;52(6):413–21.
Cordeddu V, et al. Activating mutations affecting the Dbl homology domain of SOS2 cause Noonan syndrome. Hum Mutat. 2015;36(11):1080–7.
Viskochil D. Genetics of neurofibromatosis 1 and the NF1 gene. J Child Neurol. 2002;17(8):562–70 discussion 571–2, 646–51.
Arafeh R, et al. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet. 2015;47(12):1408–10.
Flex E, et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet. 2014;23(16):4315–27.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–74.
• Jeyabalan N, Clement JP. SYNGAP1: mind the gap. Front Cell Neurosci. 2016;10:32. This is a current through review on the RasGAP protein SynGAP.
Hamdan FF, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360(6):599–605.
Hamdan FF, et al. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry. 2011;69(9):898–901.
Komiyama NH, et al. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci. 2002;22(22):9721–32.
Muhia M, et al. Disruption of hippocampus-regulated behavioural and cognitive processes by heterozygous constitutive deletion of SynGAP. Eur J Neurosci. 2010;31(3):529–43.
Vissers LE, et al. Heterozygous germline mutations in A2ML1 are associated with a disorder clinically related to Noonan syndrome. Eur J Hum Genet. 2015;23(3):317–24.
Galliano MF, et al. A novel protease inhibitor of the alpha2-macroglobulin family expressed in the human epidermis. J Biol Chem. 2006;281(9):5780–9.
Schepens I, et al. The protease inhibitor alpha-2-macroglobulin-like-1 is the p170 antigen recognized by paraneoplastic pemphigus autoantibodies in human. PLoS One. 2010;5(8):e12250.
van Trier DC, et al. External ear anomalies and hearing impairment in Noonan Syndrome. Int J Pediatr Otorhinolaryngol. 2015;79(6):874–8.
Barnes H, et al. Tyrosine-phosphorylated low density lipoprotein receptor-related protein 1 (Lrp1) associates with the adaptor protein SHC in SRC-transformed cells. J Biol Chem. 2001;276(22):19119–25.
Craig J, et al. The LDL receptor-related protein 1 (LRP1) regulates the PDGF signaling pathway by binding the protein phosphatase SHP-2 and modulating SHP-2- mediated PDGF signaling events. PLoS One. 2013;8(7):e70432.
Nacak TG, et al. The BTB-kelch protein LZTR-1 is a novel Golgi protein that is degraded upon induction of apoptosis. J Biol Chem. 2006;281(8):5065–71.
Dhanoa BS, et al. Update on the Kelch-like (KLHL) gene family. Hum Genomics. 2013;7:13.
Piotrowski A, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet. 2014;46(2):182–7.
Kraft M, et al. Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome-like phenotype and hyperactivated MAPK signaling in humans and mice. J Clin Invest. 2011;121(9):3479–91.
Clark AM, et al. Mutational activation of the MAP3K8 protooncogene in lung cancer. Genes Chromosomes Cancer. 2004;41(2):99–108.
Shchelochkov OA, et al. Duplication of chromosome band 12q24.11q24.23 results in apparent Noonan syndrome. Am J Med Genet A. 2008;146A(8):1042–8.
Graham JM Jr, et al. Genomic duplication of PTPN11 is an uncommon cause of Noonan syndrome. Am J Med Genet A. 2009;149A(10):2122–8.
Geckinli BB, et al. Clinical report of a patient with de novo trisomy 12q23.1q24.33. Genet Couns. 2015;26(4):393–400.
Chen JL, et al. Rare copy number variations containing genes involved in RASopathies: deletion of SHOC2 and duplication of PTPN11. Mol Cytogenet. 2014;7:28.
Luo C, et al. Microduplication of 3p25.2 encompassing RAF1 associated with congenital heart disease suggestive of Noonan syndrome. Am J Med Genet A. 2012;158A(8):1918–23.
Lissewski C, et al. Copy number variants including RAS pathway genes-How much RASopathy is in the phenotype? Am J Med Genet A. 2015;167A(11):2685–90.
Yu S, Graf WD. BRAF gene deletion broadens the clinical spectrum neuro-cardio-facial-cutaneous syndromes. J Child Neurol. 2011;26(12):1593–6.
Nowaczyk MJ, et al. Deletion of MAP2K2/MEK2: a novel mechanism for a RASopathy? Clin Genet. 2014;85(2):138–46.
Risheg H, et al. Clinical comparison of overlapping deletions of 19p13.3. Am J Med Genet A. 2013;161A(5):1110–6.
• Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. This paper provides guidelines that will help standardize the criteria for the assignment of a new gene variant as being causative for a given disorder.
Acknowledgments
The authors thank patients and families for their ongoing support of research in Genomic Medicine. This work was supported in part by NIH Grant HD048502 (K.A.R.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
William E. Tidyman and Katherine A. Rauen declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical collection on Clinical Genetics.
Rights and permissions
About this article

Cite this article
Tidyman, W.E., Rauen, K.A. Expansion of the RASopathies. Curr Genet Med Rep 4, 57–64 (2016). https://doi.org/10.1007/s40142-016-0100-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-016-0100-7