
Abstract
Purpose of Review
Traumatic brain injury (TBI) is a global public health problem. It is increasingly being recognized as a progressive disease in which intrinsic pathophysiologic processes and systemic secondary insults aggravate the primary brain damage. The perioperative period is crucial in the continuum of the management of TBI. This review summarizes current anesthetic strategies to optimize TBI outcomes.
Recent Findings
Perioperative data on TBI management are limited. However, recent findings indicate that intraoperative secondary insults are common and are associated with worse outcomes after intracranial as well as extracranial surgery in patients with TBI. The choice of anesthetic agents has not been shown to impact that the neurologic outcomes are TBI.
Summary
Perioperative therapeutic goals for patients with TBI include facilitating early cerebral decompression, providing balanced anesthesia for surgery, maintaining adequate cerebral perfusion by optimizing systemic and intracranial hemodynamics, and aggressively avoiding secondary insults. Intensive multimodal monitoring of the brain provides vital information that may be helpful in individualizing therapy; such monitoring should be continued in the perioperative period, particularly during extracranial surgery in TBI patients with polytrauma. While further investigation to characterize the impact of anesthetic agents on the injured brain and their effect on clinical outcomes is awaited, perioperative management should focus on adherence to consensus guidelines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide [1]. Anesthesiologists are involved in the care of patients with TBI in various situations including resuscitation and stabilization in the emergency department, providing anesthesia for evacuation of intracranial hematoma/decompressive craniectomy/extracranial surgery, and during management on the intensive care unit (ICU). This review will focus primarily on the immediate perioperative management of adult patients with TBI undergoing surgery. While a uniform approach to treating TBI victims throughout the continuum of care appears intuitive, there are challenges that are unique to the various patient care locations. Irrespective, the overall goal of the perioperative management of TBI is to minimize further brain damage, prevent secondary insults, and institute potential neuroprotective interventions. The anesthetic plan should be individualized and cogent with overall treatment goals.
Pathophysiology of TBI
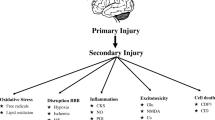
The pathology of TBI involves a primary injury that occurs at the time of the trauma and secondary injury caused by subsequent alteration of homeostasis. The initial direct trauma causes mechanical distortion, stretch and disruption of cerebral tissue resulting in focal or global alterations in cerebral autoregulation, blood flow (CBF), and metabolism [2–4]. Secondary insults such as hypotension and hypoperfusion cause further brain damage and contribute to worse outcomes [5, 6]. Brain trauma can be viewed as an acute cerebral energy crisis followed by a chronic energy shortage. The initial injury causes a release of glutamate that activates N-methyl-d aspartate receptors causing cellular efflux of potassium and influx of sodium and calcium. The adenosine triphosphate (ATP)ase pump quickly consumes ATP, depleting cells of energy in the setting of disruption of supply of oxygen and metabolic substrate [7]. A reactive global increase of cerebral glucose uptake is followed by a decrease in glucose uptake, particularly in the penumbral area surrounding the initial injury [8, 9]. The depletion of ATP and energy results in an increase in the production of free radicals and decrease in the antioxidant capacity of cells. Under normal circumstances, mitochondria transport electrons across the transport chain, but this mechanism breaks down following injury leading to a cellular energy crisis. The mitochondria are also unable to handle excess calcium and activate multiple enzymes capable of producing more free radicals [10].
The vascular derangement in TBI is a consequence of shearing and mechanical forces causing local or global abnormalities. Damage to the endothelial lining of cerebral vessels, decreased amounts of nitric oxide and cholinergic neurotransmitters, and an increase of prostaglandin induce vasoconstriction leading to decreased CBF and ischemia. Reduced CBF following TBI can result in focal or regional ischemia, which can be a portent of poor outcome [11, 12]. The key to prevention/avoidance of a cerebral ischemic cascade leading to worsening brain damage is the maintenance of adequate CBF. Consequently, cerebral hypoperfusion and hypoxia should be aggressively avoided in the perioperative period to minimize secondary brain damage.
Perioperative Management of TBI
Instead of being considered an isolated traumatic event, TBI is increasingly recognized as a progressive disease in which intrinsic pathophysiologic processes and systemic secondary insults aggravate the primary brain damage. It is now well accepted that the initial injury may evolve into a chronic disease. Consequently, the perioperative period is crucial in the continuum of the management of TBI. While surgery and anesthesia may predispose patients to new onset secondary injuries which may contribute adversely to outcomes, the perioperative period is also an opportunity to detect and correct undiagnosed pre-existing secondary insults and to prevent new insults [13]. The clinical management of TBI is largely guided by the recommendations of the Brain Trauma Foundation, which are summarized in Table 1 [14]. Adherence to guidelines in the perioperative period has been shown to be associated with decreased mortality and improved outcomes [15••, 16]. Despite the fact that these guidelines are evidence-based and have substantially improved the standards of TBI care, they do not account for the heterogeneity of TBI. In fact, it appears that guidelines have facilitated uncritical adoption of standardized approaches to TBI management without accounting for individual variation. While the adoption of guidelines is expected to reduce mortality overall, further improvement in functional outcome will likely require identification of targets for precision medicine and individualized care. Neuroimaging and biomarkers will likely contribute to better phenotyping and the development of more precise (individualized) treatment strategies. For perioperative TBI management, this might have implications in terms of individualized physiological/biochemical targets as well as administration of potential neuroprotective therapies. Meanwhile, current management of TBI emphasizes the following [14]:
-
Targeted management of cerebral perfusion pressure (CPP), mean arterial blood pressure (MAP), and intracranial pressure (ICP);
-
Maintenance of normoglycemia, normothermia, and oxygenation;
-
Prevention of secondary injury and supporting overall recovery from concomitant extracranial injuries.
Anesthetic Agents for Traumatic Brain Injury
The intravenous and inhaled anesthetic agents differ significantly in their effects on cerebral physiology. In general, volatile anesthetic agents cause a dose-dependent uncoupling of CBF and cerebral metabolic rate for oxygen (CMRO2) leading to cerebral vasodilation and increased ICP [17–20]. At concentrations <1.0 MAC, the cerebral vasodilatory effects of inhaled anesthetics are minimal, and their vasodilating effects can also be blunted by increases in minute ventilation. The intravenous agents, including thiopental, propofol, and etomidate, cause cerebral vasoconstriction and reduce cerebral blood volume (CBV) and ICP, while preserving the coupling of CBF and CMRO2 [19–23]. However, the electroencephalographic burst-suppression induced by propofol after TBI does not significantly change brain tissue oxygen levels, tissue chemistry, or arterial–jugular venous oxygen differences and does not appear to reduce the regional ischemic burden [24]. While cerebrovascular pressure autoregulation is preserved with propofol infusion in healthy individuals, high-dose propofol (plasma target concentration of ~4.3 mcg/mL) may worsen static cerebral autoregulation in patients with TBI thereby increasing vulnerability to secondary insults [25].
Importantly, the choice of anesthetic agents (inhalation vs. Intravenous) has not been shown to impact outcomes after TBI. A retrospective study did not find an association between type of anesthetic agent, i.e., intravenous or inhaled, and neurologic outcome [26]. Half the patients who received intravenous anesthesia in this series received ketamine, and there was also no difference in outcome among these patients. In the absence of conclusive evidence, either inhaled or intravenous anesthesia may be employed judiciously after TBI. A balanced anesthetic technique, including analgesics and neuromuscular blocking agents, is the best, and the principles of anesthetic management should adhere to the current guidelines for the management of severe TBI [14].
Perioperative Monitoring
In additional to standard American Society of Anesthesiology (ASA) monitoring (electrocardiography, noninvasive blood pressure, pulse oximetry, capnography, and temperature), arterial catheterization is recommended to allow beat-to-beat blood pressure, arterial blood gas, and blood glucose monitoring. ICP monitoring is recommended in patients with a severe TBI and abnormal CT scan (hematomas, contusions, swelling, herniation, or compressed basal cistern), and in patients with severe TBI and a normal CT scan if two or more of the following are present: age >40 years, motor posturing, or systolic blood pressure <90 mmHg [14]. It is generally recommended that ICP should be maintained <20 mmHg and CPP 50–70 mmHg [14]. However, the role of ICP monitoring in improving neurological outcomes has recently been questioned. A randomized controlled trial assigning severe TBI patients to therapy directed either by ICP monitoring or clinical examination and imaging reported no difference in the primary outcome of a composite of survival time, impaired consciousness, and functional status at 3 and 6 months between these groups [27]. A subsequent systematic review and meta-analysis concluded that current clinical evidence indicates that ICP monitoring-guided treatment of TBI has no mortality benefit compared to treatment in the absence of ICP monitoring [28]. Recent guidance recommends that there is no indication for ICP monitoring in comatose TBI patients with a normal computed tomography (CT) scan, but that ICP monitoring should be undertaken in comatose TBI patients with cerebral contusions in whom clinical examination cannot be performed due to sedation [29]. It remains widely accepted that ICP management has the potential to influence outcome, particularly when care is targeted, individualized, and supplemented with data from other monitors [30].
Monitoring cerebral oxygenation (global or focal), CBF or metabolic parameters may be helpful in guiding treatment decisions after TBI [16]. In TBI patients with polytrauma, all monitoring should be continued during extracranial surgery. Jugular venous oximetry has been used to detect intraoperative cerebral desaturation and guide anesthetic interventions, such as optimizing hyperventilation therapy, management of CPP, fluids and oxygenation, in order to optimize cerebral physiology [31–33]. Jugular venous saturation ≤55 % has been shown to be associated with poor neurological outcome and should be avoided [34]. Continuous brain tissue oxygen monitoring (PtiO2) reflects both oxygen delivery and consumption. The normal PtiO2 ranges from 20 to 35 mmHg, and the ischemic threshold from 10 to 15 mmHg; and values below 10 mmHg are associated with poor outcomes after TBI [35, 36]. Low brain PtiO2 can be normalized by several strategies aimed at increasing cerebral oxygen delivery. Increasing CPP can improve brain PtiO2 when cerebral autoregulation is impaired and the baseline PtiO2 is low [35]. PtiO2-guided therapy has also been linked to improved patient outcomes [36], although there are no data demonstrating the utility of PtiO2 monitoring during surgery and anesthesia for TBI.
Measurement of the CBF velocity using Transcranial Doppler (TCD) ultrasonography can provide real-time information about changes in the cerebral circulation. TCD has been used to assess CBF during extracranial surgery in TBI patients, and to examine cerebral autoregulation and guide management of systemic physiological parameters [37]. Efforts to use TCD to estimate the CPP noninvasively have not been successful.
Perioperative Hemodynamic Management
Most TBI patients present initially with a high sympathetic tone and hypertension as the first response to the trauma. This can be followed by hypotension due to shock, administration of medications to treat cerebral hypertension such as Mannitol, or medications given to facilitate intubation and airway management. The recommendation of the Brain Trauma Foundation is to maintain systolic blood pressure >90 mmHg and avoid CPP <50 mmHg [14]. When presenting for emergent evacuation of intracranial hematoma, most patients do not have an ICP monitor in place, and reliance on systolic blood pressure to guide management ignores the main driving force behind CPP, i.e., the MAP. From a practical perspective, maintaining MAP >80 mmHg in a patient requiring acute cerebral decompression is likely to ensure CPP >50 mmHg. Hypotension during emergent craniotomy for TBI occurs in 32–65 % patients and may be associated with increased hospital mortality [38, 39]. The risk factors for intraoperative hypotension are low admission Glasgow Coma Scale (GCS) score, preoperative tachycardia and hypertension, the presence of a subdural hematoma (SDH) or multiple cranial CT lesions, maximum thickness of the CT lesion and longer duration of anesthesia [39]. Specifically, sudden decrease in blood pressure often occurs following removal of the bone flap and incision of the dura, which is thought to result from sudden loss of the Cushing’s response [40]. This “decompression hypotension” may be predicted by low GCS score, the absence of mesencephalic cisterns on CT scan, and bilateral dilated pupils [40]. Intraoperative hypotension has recently been reported to occur in 60 % of TBI patients undergoing surgery for an associated orthopedic injury, and the resulting cerebral hypoperfusion was noted to lead to postoperative escalation of care [41••]. Essentially, intraoperative hypotension is common and potentially detrimental and, hence, must be avoided.
Normovolemia is the fluid resuscitation goal after TBI. In general, warm, nonglucose containing isotonic crystalloid solutions are preferable. Hypertonic saline solutions (HTS) increase intravascular volume while decreasing cerebral volume and ICP, and have other potential beneficial effects including relaxation of smooth muscle, liberation of prostacyclin from endothelial cells, vasodilation and liberation of endothelial relaxing factor [42–44]. In a post hoc analysis of trauma patients, those who received HTS in the prehospital period had higher blood pressures, decreased overall fluid requirements, and improved survival [45]. In the TBI subgroup of this study, statistically significant improvement at discharge was noted in those who received HTS. It remains to be elucidated whether the effects of HTS are due to improved MAP, innate systemic effects, or both. The controversy generated by the Saline versus Albumin Fluid Evaluation (SAFE) study [46], which demonstrated that resuscitation with albumin is associated with higher mortality rate and unfavorable neurological outcome at 24 months after TBI, still keeps many from using albumin, despite arguments refuting those findings. However, no guidelines currently recommend the use of albumin after TBI [47]. The concerns about systemic adverse effects of starches and other colloids have also severely curtailed their use, and they too do not appear in any guidelines for the management of TBI.
Treatment with vasopressors to augment blood pressure can be instituted until normovolemia is achieved, or if adequate MAP cannot be maintained despite normovolemia. The choice of vasopressor to augment CPP in TBI remains controversial. In the United States, the pure alpha-adrenergic agent phenylephrine is used most frequently, followed by norepinephrine and dopamine [48]. While some studies demonstrate a superior increase in blood pressure with phenylephrine, at least one retrospective study points to improved cardiac function with norepinephrine that might result in beneficial outcome effects after TBI [49]. Since TBI-related cardiac dysfunction is common, the beneficial effects of norepinephrine on cardiac function lead some authorities to recommend that it should be the vasopressor of choice after TBI. Additionally, norepinephrine results in a more consistent increase in the CBF velocity and cerebrovascular resistance than dopamine [50]. In a study using positron emission tomography, CMRO2 decreased when norepinephrine was used to increase CPP from 70 to 90 mmHg with [51]. One might extrapolate from this study that norepinephrine may increase CBF while decreasing consumption of oxygen, placing the traumatized area in a more favorable metabolic state. Anesthetic agents typically decrease systemic vascular resistance and MAP, so anesthesiologists should anticipate the need for vasopressors to augment blood pressure even in patients who arrive to the operating room without prior vasopressor need. The choice of vasopressor should take into account ICP, existence of cardiac dysfunction, coronary disease, and heart rate. Importantly, the arterial line should be zeroed at the level of the external auditory meatus (roughly at the level of the Circle of Willis), since accurate calculation of CPP requires that its components (MAP and ICP) are ‘zeroed’ at the same level.
Management of Intracranial Hypertension
The therapeutic options for reducing increased ICP include the following:
-
(a)
Adequate anesthesia, analgesia, and neuromuscular blockade: Inadequate sedation/analgesia can potentially increase ICP due to the increased CBF associated with increased cerebral metabolic activity. Neuromuscular blockade prevents coughing or fighting the ventilator which can also increase ICP.
-
(b)
Positioning: Careful positioning, including avoidance of obstruction to cerebral venous drainage by preventing excessive flexion and/or lateral rotation of the neck, is crucial.
-
(c)
Controlled ventilation, judicious use of hyperventilation, and maintenance of hemodynamic stability: Prophylactic/prolonged and excessive hyperventilation may worsen cerebral ischemia and must be avoided.
-
(d)
Hyperosmolar therapy: Both Mannitol and hypertonic saline are effective treatments of TBI-related intracranial hypertension. Mannitol (0.25–1.0 g/kg) reduces ICP by two distinct mechanisms—a reflex vasoconstriction caused by viscosity reduction leading to improved microvascular perfusion, and an osmotic diuretic effect. Hypertonic saline also has favorable rheologic and osmolar gradient effects but may offer other potential beneficial actions including restoration of normal cellular resting membrane potential and cell volume, inhibition of inflammation, and enhancement of cardiac output (see above).
-
(e)
Cerebrospinal fluiddrainage: An external ventricular drain (EVD) allows for a continuous monitoring of ICP and treatment of increases in ICP by allowing drainage of cerebral spinal fluid.
Glycemic Management
Hyperglycemia can cause secondary brain damage and has been shown to be associated with worse outcomes following TBI [52, 53]. However, tight glycemic control can increase cerebral metabolic crisis [53] and, interestingly, in vitro and in vivo studies suggest a direct benefit from increased glucose levels during the acute phase after TBI [54•, 55–57]. It is clear that glucose metabolism is disrupted in TBI and that the body tries to increase the availability of this important substrate. Hence, tight glucose control with intensive insulin therapy remains controversial, and may be undesirable in the intraoperative period when glucose values may fluctuate widely. Nevertheless, intraoperative hyperglycemia has been reported to occur in 15–20 % adults undergoing urgent/emergent craniotomy for TBI, and can be predicted by severe TBI, the presence of SDH, preoperative hyperglycemia, and aged ≥65 years [58, 59•]. Given the current evidence for glucose control for TBI in the perioperative period, a target glucose range of 80–140 mg/dl appears reasonable.
Hypothermia in TBI
Extensive research has explored the use of hypothermia as a therapeutic strategy to decrease ICP and improve neurological outcomes after TBI. Therapeutic cooling of the brain is postulated to attenuate secondary brain injury due to amelioration of damaging inflammatory and neuroexcitatory cascades and elevated ICP [60–62]. Despite promising results in preclinical studies, the results of clinical studies are not encouraging and recent trials of hypothermia indicate a potential for clinical harm. The Eurotherm3235 Trial was a large randomized, controlled trial that tested therapeutic hypothermia as the primary intervention to reduce ICP after TBI when first-line ICP controlling therapies failed [63••]. In this trial involving patients with ICP >20 mm Hg for at least 5 min, despite first-line interventions (sedation, analgesia, mechanical ventilation, and head elevation), hypothermia plus standard care did not result in outcomes that were superior to those with standard care alone. Moreover, 36.5 % patients in the standard care group achieved a favorable outcome compared with 25.7 % in the hypothermia arm of the trial (p = 0.03), and the mortality in the hypothermia group was higher than that in the normothermia group (34.9 % vs. 26.6 %, hazard ratio, 1.45: 95 % CI 1.01–2.10; p = 0.047). The study was terminated early because of the adverse effects of hypothermia. In the perioperative period, particularly during anesthesia, patients are likely to become hypothermic but this is unlikely to provide any therapeutic benefit. In fact, hypothermia during acute bleeding may contribute to worsening coagulopathy and should be avoided.
Anesthesia for Extracranial Surgery
Patients with TBI often have associated other injuries which may require surgery, such as orthopedic, abdominal, and/or thoracic surgery. The timing and extent of the extracranial surgery depend on TBI severity as well as the severity of the extracranial injuries [64]. Importantly, the recovery of cerebral autoregulation after severe TBI can be delayed for weeks, particularly in patients with a lower GCS score, diffuse brain injury, and elevated ICP [65], and those with delayed recovery of autoregulation tend to have a worse outcome. Moreover, patients with traumatic subarachnoid hemorrhage are also at risk of cerebral vasospasm for 2–15 days after TBI, and this can exacerbate the risk of cerebral ischemia [66]. Consequently, the brain is susceptible to further ischemic damage even weeks after the original injury. Although hypotension should obviously be avoided during extracranial surgery after TBI, it has been reported in up to 60 % of adult cases and up to 78 % of pediatric cases [41, 67]. Not surprisingly, cerebral hypotension during extracranial surgery leads to escalation of postoperative care and lower chance of good neurological outcome. Other secondary insults, such as inadvertent hypercarbia and hypocarbia, are also common during extracranial surgery and potentially detrimental [41, 67].
The principles of anesthetic management of patients with TBI presenting for noncranial surgery are largely the same as those for craniotomy. Importantly, all preoperative monitoring should be continued during transport to the operating room and during surgery. In patients receiving antiepileptic medications, drug levels should be checked and administration continued uninterrupted perioperatively since seizures can increase ICP and cause further metabolic derangement [68].
Anesthesia should be induced carefully, avoiding hemodynamic fluctuations, hypoxia, and hypercarbia. While intravenous anesthesia with propofol may have some advantage in terms of lower ICP, there are no good data to support the use of intravenous over inhaled anesthetic agents. Increased intraoperative ICP due to inadvertent hypercarbia and improper positioning must be avoided. ICP should be closely monitored and the anesthesiologist prepared to treat intraoperative intracranial hypertension urgently.
Future Directions
There is paucity of perioperative, especially intraoperative, data on TBI management. Limited data also exist on the effect of anesthetic agents on the histopathological features of TBI, and further investigation is warranted to better characterize the impact of anesthetic agents on the injured brain. Anesthesiologists will need to partner with other disciplines to conduct larger, multi-centric studies to better guide anesthetic management. In order to formulate detailed clinical management for the very dynamic perioperative period, such studies will need to characterize the injury itself, bear in mind that TBI is a heterogeneous disease process, and collect imaging and biomarker data in addition to anesthetic details. Common data elements for research into the intraoperative TBI management have been developed [69], and future studies should incorporate them to allow uniform reporting as well as potential pooling of data. Meanwhile, perioperative management of TBI should focus on adherence to multidisciplinary, consensus guidelines.
Conclusion
The perioperative period is crucial in the continuum of TBI management. Perioperative management goals include facilitating early cerebral decompression, providing balanced anesthesia for surgery, maintaining adequate cerebral perfusion by optimizing systemic and intracranial hemodynamics, and aggressively avoiding secondary insults. Multimodal neuromonitoring should be continued in the perioperative period, particularly during extracranial surgery in TBI patients with polytrauma. While further investigation to characterize the impact of anesthetic agents on the injured brain and their effect on clinical outcomes is awaited, perioperative management should focus on adherence to multidisciplinary, consensus guidelines.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hyder AA, Wunderlich CA, Puvanachandra P, et al. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–53.
Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–9.
Cunningham AS, Salvador R, Coles JP, et al. Physiological thresholds for irreversible tissue in contusional regions following brain injury. Brain. 2005;128:1931–42.
Bouma GJ, Muizelaar JP, Stringer WA, et al. Ultra-early evaluation of regional cerebral blood flow in severe head-injured patients using Xenon-enhanced computerized tomography. J Neurosurg. 1992;77:360–8.
McHugh G, Engel D, Esteyengerg E, et al. Prognostic value of secondary insults in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24(2):287–93.
Chesnut RM, Marshall LF, Klauber MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216–22.
Katayama Y, Becker DP, Tamura T, et al. Massive increases in extracelluar potassium and the indiscriminate release of glutamate following contusive brain injury. J Neurosurg. 1990;7:889–900.
Bergsneider MA, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe human traumatic brain injury: a positron emission tomography study. J Neurosurg. 1997;86:241–51.
Vespa P, McArthur D, Glenn T, et al. Persistently reduced levels of extracellular glucose early after traumatic brain injury correlate with poor outcome at six months: a microdialysis study. J Cereb Blood Flow Metab. 2003;23:865–77.
Xiong Y, Gu Q, Peterson PL, et al. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J Neurotrauma. 1997;14:23–34.
Coles JP, Fryer TD, Smielewski P, et al. Incidence and mechanism of cerebral ischemia in early clinical head injury. J Cereb Flow Metab. 2004;24:202–11.
Inoye Y, Shiozaki T, Tasaki O, et al. Changes in cerebral blood flow from the acute to the chronic phase of severe head injury. J Neurotrauma. 2005;22:1411–8.
Curry P, Viernes D, Sharma D. Perioperative management of traumatic brain injury. Int J Crit Illn Inj Sci. 2011;1:27–35.
Brain TF. Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24(Suppl 1):S1–106.
•• Gupta D, Sharma D, Kannan N, et al. Guideline adherence and outcomes in severe adult traumatic brain injury for the CHIRAG (Collaborative Head InjuRy and Guidelines) study. World Neurosurg. 2016;89:169–79 [Epub ahead of print] This study demonstrates improved outcomes in severe TBI in India when the rate of adherence to established guidelines are improved.
Lee JC, Rittenhouse K, Bupp K, et al. An analysis of Brain Trauma Foundation traumatic brain injury guideline compliance and patient outcome. Injury. 2015;46:854–8.
Oshima T, Karasawa F, Okazaki Y, et al. Effects of sevoflurane on cerebral blood flow and cerebral metabolic rate of oxygen in human beings: a comparison with isoflurane. Eur J Anaesthesiol. 2003;20:543–7.
Kaisti KK, Långsjö JW, Aalto S, et al. Effects of sevoflurane, propofol, and adjunct nitrous oxide on regional cerebral blood flow, oxygen consumption, and blood volume in humans. Anesthesiology. 2003;99:603–13.
Matta BF, Mayberg TS, Lam AM. Direct cerebrovasodilatory effects of halothane, isoflurane, and desflurane during propofol-induced isoelectric electroencephalogram in humans. Anesthesiology. 1995;83:980–5.
Oshima T, Karasawa F, Satoh T. Effects of propofol on cerebral blood flow and the metabolic rate of oxygen in humans. Acta Anaesthesiol Scand. 2002;46:831–5.
Petersen KD, Landsfeldt U, Cold GE, et al. Intracranial pressure and cerebral hemodynamic in patients with cerebral tumors: a randomized prospective study of patients subjected to craniotomy in propofol-fentanyl, isoflurane-fentanyl, or sevoflurane-fentanyl anesthesia. Anesthesiology. 2003;98:329–36.
Pinaud M, Lelausque JN, Chetanneau A, et al. Effects of propofol on cerebral hemodynamics and metabolism in patients with brain trauma. Anesthesiology. 1990;73:404–9.
Shapiro HM, Galindo A, Wyte SR, et al. Rapid intraoperative reduction of intracranial pressure with thiopentone. 1973. Br J Anaesth. 1998;81:798–803.
Johnston AJ, Steiner LA, Chatfield DA, et al. Effects of propofol on cerebral oxygenation and metabolism after head injury. Br J Anaesth. 2003;91:781–6.
Steiner LA, Johnston AJ, Chatfield DA, et al. The effects of large-dose propofol on cerebrovascular pressure autoregulation in head-injured patients. Anesth Analg. 2003;97:572–6.
Grathwohl KW, Black IH, Spinella PC, et al. Total intravenous anesthesia including ketamine versus volatile gas anesthesia for combat-related operative traumatic brain injury. Anesthesiology. 2008;109:44–53.
Chesnut RM, Temkin N, Carney N, et al. A trial of intracranial-pressure monitoring in traumatic brain injury. N Engl J Med. 2012;367:2471–81.
Yuan Q, Wu X, Sun Y. Impact of intracranial pressure monitoring on mortality in patients with traumatic brain injury: a systematic review and meta-analysis. J Neurosurg. 2014;5:1–14.
Stocchetti N, Picetti E, Berardino M, et al. Clinical applications of intracranial pressure monitoring in traumatic brain injury: report of the Milan consensus conference. Acta Neurochir (Wien). 2014;156:1615–22.
Chesnut R, Videtta W, Vespa P, et al. Intracranial pressure monitoring: fundamental considerations and rationale for monitoring. Neurocrit Care. 2014;21(Suppl 2):S64–84.
Chan KH, Miller JD, Dearden NM, et al. The effect of changes in cerebral perfusion pressure upon middle cerebral artery blood flow velocity and jugular bulb venous oxygen saturation after severe brain injury. J Neurosurg. 1992;77:55–61.
Skippen P, Seear M, Poskitt K, et al. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med. 1997;25:1402–9.
Sharma D, Siriussawakul A, Dooney N, et al. Clinical experience with intraoperative jugular venous oximetry during pediatric intracranial neurosurgery. Paediatr Anaesth. 2013;23:84–90.
Pérez A, Minces PG, Schnitzler EJ, et al. Jugular venous oxygen saturation or arteriovenous difference of lactate content and outcome in children with severe traumatic brain injury. Pediatr Crit Care Med. 2003;4:33–8.
De Georgia MA. Brain tissue oxygen monitoring in neurocritical care. J Intensive Care Med. 2015;30:473–83.
Narotam PK, Morrison JF, Nathoo N. Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg. 2009;111:672–82.
Bouzat P, Oddo M, Payen JF. Transcranial doppler after traumatic brain injury: is there a role? Curr Opin Crit Care. 2014;20:153–60.
Kinoshita K, Kushi H, Sakurai A, et al. Risk factors for intraoperative hypotension in traumatic intracranial hematoma. Resuscitation. 2004;60:151–5.
Sharma D, Brown MJ, Curry P, et al. Prevalence and risk factors for intraoperative hypotension during craniotomy for traumatic brain injury. J Neurosurg Anesthesiol. 2012;24:178–84.
Kawaguchi M, Sakamoto T, Ohnishi H, et al. Preoperative predictors of reduction in arterial blood pressure following dural opening during surgical evacuation of acute subdural hematoma. J Neurosurg Anesthesiol. 1996;8:117–22.
•• Algarra NN, Lele AV, Prathep S, et al. Intraoperative Secondary Insults During Orthopedic Surgery in Traumatic Brain Injury. J Neurosurg Anesthesiol. 2016, Mar 4 [Epub ahead of print] This study is one of the first one to look at the intraoperative period as one of high importance during the hospital treatment of TBI. The types of secondary injuries and the rate of incidence are highlighted as should serve as call to action to all who care for TBI patients in the operating rooms.
Schmoker JD, Shackford SR, Wald SL, et al. An analysis of the relationship between fluid and sodium administration and intracranial pressure after head injury. J Trauma. 1992;33:476–81.
Hartl R, Schurer L, Schmid-Schonbein GW, et al. Experimental antileukocyte interventions in cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:1108–19.
Hartl R, Medary MB, Ruge M, et al. Hypertonic/hyperoncotic saline attenuates microcirculatory disturbances after traumatic brain injury. J Trauma. 1997;42:S41–7.
Vassar MJ, Perry CA, Gannaway WL, Holcroft JW. 7.5% sodium chloride/dextran for resuscitation of trauma patients undergoing helicopter transport. Arch Surg. 1991;1(126):1065–72.
Myburgh J, Cooper DJ, Finfer S, et al. Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;2007(357):874–84.
Van Aken HK, Kampmeier TG, Ertmer C, et al. Fluid resuscitation in patients with traumatic brain injury: what is a SAFE approach? Curr Opin Anesthesiol. 2012;25:563–5.
Sookplung P, Siriussawakul A, Malakouti A, et al. Vasopressor use and effect on blood pressure after severe adult traumatic brain injury. Neurocrit Care. 2011;15:46–54.
Krishnamoorthy V, Prathep S, Sharma D, et al. Association between electrocardiographic findings and cardiac dysfunction in adult isolated traumatic brain injury. Indian J Crit Care Med. 2014;18:570–4.
Steiner LA, Johnston AJ, Czosnyka M, et al. Direct comparison of cerebrovascular effects of norepinephrine and dopamine in head-injured patients. Crit Care Med. 2004;32:1049–54.
Steiner LA, Coles JP, Johnston AJ, et al. Assessment of cerebrovascular autoregulation in head-injured patients a validation study. Stroke. 2003;34:2404–9.
Young B. Relationship between admission hyperglycemia and neurologic outcome of severely brain injured patients. Ann Surg. 1989;210:466–73.
Lam A. Hyperglycemia and neurological outcome in patients with head injury. J Neurosurg. 1991;75:545–51.
• Vespa P, McArthur DL, Stein N, et al. Tight glycemic control increases metabolic distress in traumatic brain injury: A randomized controlled within-subjects trial.Crit Care Med. 2012;40:1923–29. This important study adds to the current view that tight glycemic control in the TBI patient is not advisable and could be detrimental as the brain tries to “self-regulate” substrate consumption vs supply.
Seo SY, Kim EY, Kim H, et al. Neuroprotective effect of high glucose against NMDA, free radical, and oxygen-glucose deprivation through enhanced mitochondrial potentials. J Neurosci. 1999;19:8849–55.
Ginsberg MD, Prado R, Dietrich WD, et al. Hyperglycemia reduces the extent of cerebral infarction in rats. Stroke. 1987;18:570–4.
Zasslow MA, Pearl RG, Shuer LM, et al. Hyperglycemia decreases acute neuronal ischemic changes after middle cerebral artery occlusion in cats. Stroke. 1989;20:519–23.
Pecha T, Sharma D, Hoffman NG, et al. Hyperglycemia during craniotomy for adult traumatic brain injury. Anesth Analg. 2011;113:336–42.
• Bhattacharjee S, Layek A, Maitra S, et al. Perioperative glycemic status of adult traumatic brain injury patients undergoing craniotomy: a prospective observational study. J Neurosurg Anesthesiol. 2014;26:313–9. This study further elucidates the derangements in glucose metabolism associated with TBI. Even in patients without previous metabolic disorders hyperglycemia can be present and exacerbated by medical interventions.
Chatzipanteli K, Alonso OF, Kraydieh S, et al. Importance of posttraumatic hypothermia and hyperthermia on the inflammatory response after fluid percussion brain injury: biochemical and immunocytochemical studies. J Cereb Blood Flow Metab. 2000;20:531–42.
Clifton GL, Jiang JY, Lyeth BG, et al. Marked protection by moderate hypothermia after experimental traumatic brain injury. J Cereb Blood Flow Metab. 1991;11:114–21.
Sutcliffe IT, Smith HA, Stanimirovic D, et al. Effects of moderate hypothermia on IL-1 beta-induced leukocyte rolling and adhesion in pial microcirculation of mice and on proinflammatory gene expression in human cerebral endothelial cells. J Cereb Blood Flow Metab. 2001;21:1310–9.
•• Andrews PJ, Sinclair HL, Rodriguez A, et al; Eurotherm3235 Trial collaborators. hypothermia for intracranial hypertension after traumatic brain injury. N Engl J Med. 2015;373:2403–12 This much awaited study did not show any improvement with hypothermia therapy vs standard of care and seems to follow the current Brain Trauma Foundation recommendations as we adapt them to the operating rooms.
Velmahos GC, Arroyo H, Ramicone E, et al. Timing of fracture fixation in blunt trauma patients with severe head injuries. Am J Surg. 1998;176:324–9.
Sviri GE, Aaslid R, Douville CM, et al. Time course for autoregulation recovery following severe traumatic brain injury. J Neurosurg. 2009;111:695–700.
Razumovsky A, Tigno T, Hochheimer SM, et al. Cerebral hemodynamic changes after wartime traumatic brain injury. Acta Neurochir Suppl. 2013;115:87–90.
Fujita Y, Algarra NN, Vavilala MS, Prathep S, Prapruettham S, Sharma D. Intraoperative secondary insults during extracranial surgery in children with traumatic brain injury. Child’s Nervous System. 2014;1(30):1201–8.
Vespa PM, Nuwer MR, Nenov V, Ronne-Engstrom E, Hovda DA, Bergsneider M, Kelly DF, Martin NA, Becker DP. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg. 1999;91(5):750–60.
https://commondataelements.ninds.nih.gov/tbi.aspx#tab=Data_Standards.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Nelson Nicolas Algarra and Deepak Sharma declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical collection on Neuroanesthesia.
Rights and permissions
About this article

Cite this article
Algarra, N.N., Sharma, D. Perioperative Management of Traumatic Brain Injury. Curr Anesthesiol Rep 6, 193–201 (2016). https://doi.org/10.1007/s40140-016-0170-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40140-016-0170-9