
Abstract
Liver cancer, particularly hepatocellular carcinoma (HCC), is the third leading cause of cancer death in the world. Bile acids (BAs) are liver-produced amphipathic molecules that are required to facilitate the absorption of cholesterol, fat-soluble vitamins, and lipids in the intestine. However, BAs are also known to act as potential carcinogens and deregulation of BA homeostasis has been linked to HCC formation. Two key BA receptors, farnesoid X receptor (FXR) and G protein-coupled bile acid receptor 1 (TGR5), were recently identified, which provides great insights into BAs’ normal physiological functions as well as their carcinogenic effects. In this review, we focus on the potential links among BAs, two BA receptors, and HCC. FXR and TGR5 not only play key roles in regulating BA homeostasis but also are essential in suppressing BAs’ carcinogenic effects on liver cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bile acids (BAs) are a group of water-soluble steroids formed after the catabolism of cholesterol, and synthesized in hepatocytes of the liver [1]. There are two primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA). After being dehydroxylated by the intestinal bacteria, these two primary BAs are then transformed into secondary BAs. CA becomes deoxycholic acid (DCA) and CDCA becomes lithocholic acid (LCA). Thereafter, BAs are reabsorbed by the intestine and then circulated back to the liver, where BAs are further conjugated with glycine or taurine. The recycling of BAs from intestine back to liver is known as the enterohepatic circulation [2].
The levels of BAs must be tightly controlled. Deregulation of BAs leads to several pathological consequences, such as cholestasis and cancer. Since 1940, studies have suggested that BAs could act as potential carcinogens [3]. Abnormally high concentrations of BAs induce cell death and inflammation, thereby promoting cancer development. Due to its high levels in the gastro-intestinal (GI) tract, BAs have been highly linked to the risk of GI cancers, including liver cancer.
Bile Acids and Liver Cancer
Liver cancer, particularly hepatocellular carcinoma (HCC), is the third leading cause of cancer death in the world [4]. Many studies have shed light on the initiation and development of HCC. HCC is usually associated with the chronic inflammation in liver, including the infection of hepatitis B or C virus and non-alcoholic steatohepatitis (NASH) [5]. The liver is the largest organ for metabolism and detoxification. Therefore, both xenobiotics and endobiotics, such as BAs, may induce liver injury. Inside the liver, BAs are useful to promote bile flow and eliminate cholesterol, bilirubin, steroid hormone derivatives, and xenobiotics [6]. However, abnormally high levels of BAs induce hepatocyte DNA damage, which could significantly increase the mutation rate of tumor suppressor genes and oncogenes. Furthermore, BAs can induce cell death and inflammation to promote carcinogenesis. The association of BAs with liver cancer has been demonstrated from both human and animal studies. High levels of BAs in the liver will result in cholestasis and the link between cholestatic liver diseases and liver cancer has been established in clinical studies [7]. Knisely et al. [8••] reports a link between progressive familial intrahepatic cholestasis type 2 and HCC in children who have cholestasis that results in accumulation of BAs in the liver. Mutations in the bile salt export pump, ABCB11, also result in liver tumor formation [9]. These results strongly suggest that BAs may directly contribute to the development of HCC in humans. Moreover, these children are also prone to developing cholangiocarcinoma, an adenocarcinoma that arises from the bile duct epithelium [7]. In rats, the elevated BA concentrations play a role as an endogenous promoter in hepatocarcinogenesis [10–12]. Similarly, mice with multi-drug resistance 2 (mdr2) gene deletion have accumulation of BAs inside the liver. These mice display progressive inflammation, and then HCC and cholangiocarcinoma [13]. Feeding mice with a CA-enriched diet strongly promoted diethylnitrosamine (DEN)-initiated hepatocarcinogenesis in mice [14••]. Usually, the hydrophobic BAs, such as LCA, induce higher degree of liver injury. Whether there is a correlation between the hydrophobicity of BAs and their capacity to promote liver cancer is still unclear. Also in human, the primary BA species that induce liver cancer need to be identified and verified in the future studies.
In vitro studies show that BAs may directly induce hepatocyte death by ROS-mediated cell apoptosis [15]. Other studies indicate that BAs activate the Fas receptor in a ligand-independent manner, thereby inducing the apoptosis of primary hepatocytes [16, 17]. Interestingly, BAs simultaneously activate certain cell survival pathways, including extracellular signal-regulated kinase (ERK) pathway and AKT pathway, which can suppress cell apoptosis [18]. The phosphorylation of ERK1, ERK2, and AKT is increased after BA treatments. However, the mechanism by which BAs induce those cell protective pathways is still unknown.
It is now clear that, in addition to their important roles in facilitating nutritional absorption, BAs are also signaling molecules that activate BA receptors to regulate gene expression. Two major receptors of BAs have been identified: the nuclear receptor FXR [19, 20], and TGR5, a G protein-coupled receptor [21••, 22, 23•, 24•, 25]. Through these two receptors, BAs are able to regulate glucose and lipid metabolism and modulate the immune responses [26, 27]. On the other hand, studies on FXR and TGR5 also shed light on the roles of BAs in liver cancer.
FXR as a Bile Acid Nuclear Receptor
In 1995, FXR was first discovered from a rat liver cDNA library by Forman et al. [28••]. Rodents have two FXR isoforms, FXRα and FXRβ. In humans, FXRα is expressed, while FXRβ exists as a non-expressed pseudogene [29]. FXR is mainly expressed in the liver, gut, kidneys and adrenal glands. FXR is the primary BA receptor to maintain the homeostasis of BAs by regulating the uptake, synthesis, conjugation, and transportation of BAs in the liver and gut. Previously, we already have reviews on FXR and its roles in metabolic regulation [19, 20].
FXR and Liver Repair/Regeneration
FXR not only controls the levels of BAs, but also promotes liver regeneration in response to the increased BA stress after 70 % partial hepatectomy or liver injury [30, 31, 32•]. The unexpected role of FXR in promoting liver regeneration/repair may reflect FXR’s protective role to prevent BA-induced liver toxicity after liver resection or injury [20, 33]. The synthesis of BAs in the liver is rapidly and strongly repressed after 70 % partial hepatectomy or liver injury. To efficiently control the BA levels after liver damage, FXR not only efficiently rebuilds the BA homeostasis but also directly stimulates liver regrowth to prevent BA-induced toxicity. This is further demonstrated by the activation of FXR in both intestine and liver after liver injury. FXR in the intestine has been identified to contribute significantly to suppress BA synthesis in the liver [34•, 35]. The identification of the role of intestinal FXR in promoting liver regeneration/repair suggests that FXR may directly couple BA regulation to liver regrowth [36].
FXR and Liver Cancer
In the absence of FXR, the injured liver may fail to finish normal regeneration, leading to repeated cycles of cell necrosis and compensatory proliferation of hepatocytes. This irregular proliferation of hepatocytes is an important factor in promoting hepatocarcinogenesis [37]. Indeed, FXR-null (FXR−/−) mice spontaneously develop liver tumors as they aged [14••, 34•]. As early as mice are 3 months old, far before tumor appears, FXR−/− mice have increased expression of the proinflammatory cytokine IL-1β, the proto-oncogene β-catenin, and a β-catenin target gene c-myc [14••, 38]. The significant increase of β-catenin activation could be observed at 8-months. At 14-months stage, there is 100 % of tumor incidence rate in FXR-deficient mice, which is associated with major Wnt/β-catenin pathway activation. Feeding FXR−/− mice with a CA diet further promoted chemical-induced hepatocarcinogenesis [14••]. Zhang et al. [39] showed that in FXR−/− mice, liver carcinogenesis developed more rapidly under diabetic conditions, suggesting a promoting effect of diabetes-induced inflammation on HCC. In FXR−/− mouse livers, IFNγ is one of the most up-regulated cytokines [14••]. The role of IFNγ in HCC is still unclear [40]. A recent report shows that IFNγ deletion promoted hepatocarcinogenesis in FXR−/− mice and sensitized mice to the DEN-induced tumorigenesis. IFNγ probably suppresses HCC development in FXR−/− mice by maintaining the activation of p53 and nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in aging livers [41].
In addition to promoting liver repair, FXR may protect against hepatocarcinogenesis by modulating NF-κB-mediated hepatic inflammatory responses. NF-κB is known to mediate the effect of chronic inflammation on HCC development [13, 42••]. FXR is shown to negatively regulate NF-κB signaling. Overexpression of FXR suppresses the expression of inflammatory mediators in both HepG2 cells and the primary hepatocytes. Moreover, FXR−/− mice showed increased levels of several proinflammatory cytokines in response to lipopolysaccharide (LPS) challenge. Interestingly, FXR activation has no negative effects on NF-κB-activated anti-apoptotic genes, suggesting that FXR may selectively inhibit NF-κB-mediated hepatic inflammatory response but maintains or even enhances the cell survival response [43••].
Another potential role of FXR in suppressing inflammation may be through the activation of the cytokine inducible SH2-containing protein (CISH). CISH, a therapeutic target for inflammatory diseases, represses cytokine signaling by reducing JAK2/STAT5 activation. FXR upregulates CISH at both transcriptional and translational levels, thereby inhibiting IL-6-induced STAT5 activation [44].
Based on the results so far, FXR may confer hepatoprotection and suppress HCC at multiple levels (Table 1). First, FXR controls BA homeostasis to defend against BA-induced liver injury [14••, 33, 34•]. Second, FXR prevents hepatocyte apoptosis and liver injury [30, 45]. Third, FXR may suppress ROS generation [46]. Fourth, FXR activation lowers the sensitivity of hepatic stellate cells (HSCs) to TGFβ and represses the secretion of ECM by HSCs. Indeed, FXR expression is reduced in HSCs of fibrotic livers, which contributes to the enhanced fibrosis [47]. Fifth, FXR prevents hepatic inflammation [43••]. Sixth, upon liver damage, FXR promotes liver repair and regeneration to restore the homeostasis of liver [32•, 33, 36]. Finally, FXR activates the expression of SHP, which is also a HCC suppressor as described previously [48•]. FXR/SHP axis may represent a novel HCC suppressing pathway that is distinct from their initially identified function in BA regulation. Recent reports indicate that FXR can directly suppress liver cancer cell proliferation [49, 50], which may add another level of protection by FXR as a tumor suppressor. However, the mechanism is still unclear and the conclusion is still controversial as another report has opposite observation that FXR promotes cancer cell proliferation [51].
FXR−/− mice also offered a unique animal model to better understand human hepatocarcinogenesis. HCC generally develops from sequential pathological changes, which include inflammation, compensatory regeneration, liver matrix alteration, fibrosis and cirrhosis. Studies have shown that FXR expression is decreased in hepatitis C virus-induced liver fibrosis and human HCC samples [52]. Similarly, in human HCC samples, the expression of both FXR and its target gene SHP is significantly decreased [50, 53]. FXR expression can be downregulated by inflammatory cytokines, which are upregulated in HCC microenvironment [53]. Recently, FXR is identified as a target of miR-421 in HCC cells. miR-421 suppresses the translation of FXR protein by targeting the 3′-UTR of FXR mRNA [54]. These results suggest that FXR may be one of the downstream targets of chronic inflammation after virus infection or other factors. The functional deficiency of FXR will subsequently result in the disruption of liver metabolic homeostasis, which may constitute a second hit to further exacerbate cell death, inflammation and HCC development.
In summary, in addition to acting as a metabolic regulator, FXR is also critical to protect the liver from cancer development. FXR−/− mice recapitulate many features of human hepatocarcinogenesis. Inflammation downregulates FXR expression, thereby leading to metabolic dysfunction of liver and the development of HCC.
TGR5 as a Bile Acid Membrane-Bound Receptor
The identification of FXR as the BA receptor has greatly facilitated our understanding of BA regulation. In 2002, two independent research groups identified that BAs are also ligands for a novel G protein-coupled receptor TGR5 (M-BAR, Gpbar1) [21••, 22]. Since then, TGR5 has provided further insights into the BA function as well as their roles in pathophysiology.
TGR5 and Metabolic Regulation
TGR5 is ubiquitously expressed, but its expression varies among different tissues. It is expressed at high levels in the liver, intestine, brown adipose tissue and spleen [21••, 22, 55, 56]. In vitro studies have demonstrated that cAMP accumulation after activation of TGR5 is key to the function of TGR5 [57]. Compared to FXR, TGR5 seems to be more sensitive to hydrophobic BAs [25]. The ability of BAs to suppress diet-induced obesity is mediated by TGR5 through the activation of type 2 iodothyronine deiodinase in brown adipose and skeletal muscle tissues, which allows for fine-tuning of energy homeostasis [58–60]. The notion that TGR5 has a role in energy metabolism is further supported by TGR5 knockout (TGR5−/−) mice, which accumulate fat faster than wild-type mice [23•, 24•]. In addition, mice that were fed a high fat diet and then treated with oleanolic acid (OA), a TGR5-specific ligand, exhibited lowered serum glucose and insulin levels, suggesting that TGR5 may play a role in type 2 diabetes [61]. Moreover, activation of TGR5 induced the production of glucagon-like peptide-1 (GLP-1) in an enteroendocrine cell line STC-1 [62], which was further confirmed by using mice that over-expressed TGR5 [63•, 64]. Taken together, these studies indicate that TGR5 is a key receptor for mediating the effects of BAs in regulating energy metabolism and insulin signaling.
TGR5 and Inflammation Suppression
Another well-defined function of TGR5 is its potent effect on suppressing inflammation. TGR5 mRNA is present in resting CD14+ monocytes in fractionated human leukocytes and in adherent alveolar macrophage cells [22], indicating TGR5 may have a role in modulating inflammatory response. In THP-1 cells that over-expressed TGR5 [22], BA activation of TGR5 strongly suppressed LPS-induced cytokine production, which suggests that TGR5 is a mediator of BA-induced suppression of macrophage functions. TGR5−/− mice developed more severe liver steatosis after 8 weeks of high fat diet feeding compared to the wild-type mice. Treatment of wild-type mice with a TGR5 agonist (INT-777) could reduce liver fatty acid and triglyceride contents as well as plasma triglyceride concentrations [27]. In addition, in an animal model of multiple sclerosis, treatment with OA ameliorated neurological signs of the disease by modulating inflammation and immune responses [65]. Many types of cell are involved in chronic inflammatory disorder, and dendritic cells (DCs) are one of the antigen-presenting cells carrying out innate and acquired immune responses [66]. BAs can regulate DC differentiation through the TGR5-cAMP pathway, but not through FXR. TGR5 activation stimulates the differentiation of DCs toward IL-12 hypo-producing phenotype [67]. Similar to FXR, TGR5 also has a potent ability to suppress hepatic inflammation by antagonizing NF-κB activities [42••]. TGR5 activation prevents the phosphorylation of IκBα, reduces nuclear translocation of p65, and decreases NF-κB DNA-binding activities. TGR5−/− mice have elevated inflammatory infiltration, increased rate of apoptotic cells, and higher ALT levels after LPS treatments [42••].
TGR5 and Liver Cancer
A recent study shows that TGR5 also modulates the activation of signal transducer and activate transcription 3 (STAT3) [68••]. STAT3 is a transcription factor which plays critical role in liver inflammation and cancer [69]. Chronic higher levels of STAT3 have been frequently observed in human inflammatory diseases and cancers. TGR5 activation suppresses STAT3 phosphorylation, inhibiting its transcription and DNA binding activities. TGR5−/− mice were more sensitive to DEN-induced acute liver injury and HCC compared with wild-type mice. Expression of the proinflammatory cytokines are increased in TGR5 deficient mice, including IL-1β, TNFα, IL-6, IFN-γ, monocyte chemoattractant protein-1 (MCP-1) and interferon inducible protein-10 (IP-10). Expression of matrix metalloproteinases (MMPs) is also higher in TGR5−/− mice, which may promote the development and migration of HCC [68••].
In summary, emerging evidence indicates that the membrane BA receptor TGR5 is important to mediate several aspects of BA signaling. TGR5 plays important roles in metabolic regulation and inflammatory response. In addition, TGR5 may also be a potential tumor suppressor in liver cancer.
FXR and TGR5 as Drug Targets
Both FXR and TGR5 are attractive targets for drug development. We have previously summarized the synthetic or natural ligands for FXR and TGR5 [19, 25]. The search for more potent and selective compounds for FXR and TGR5 is still going. Several new FXR ligands are identified and some of FXR ligands are currently under clinical trials already [70–73]. The screening for new TGR5 ligands are also very active and we expect some of TGR5 ligands will enter clinical trials in the near future [74, 75]. The primary clinical trials are focused on targeting metabolic diseases such as primary biliary cirrhosis (PBC). However, with more studies on the roles of FXR and TGR5 in HCC, the application of their ligands in the prevention or treatment of liver cancer will be the next objective.
Conclusions
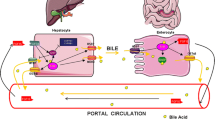
The amphiphilic characters of bile acids are required to help nutrition absorption in the small intestines, but also process potential carcinogenic effect. Therefore, BA homeostasis is not only important to help regulate other metabolic pathways associated with the diet input but also is key to prevent carcinogenic effect due to dysregulation of BA metabolism. Two BA receptors, FXR and TGR5, may coordinate each other to maintain levels of BAs and regulate BA-mediated signaling pathways. As noted, FXR and TGR5 are differentially expressed in the liver. FXR is highly expressed in hepatocytes and cholangiocytes. TGR5 is mainly expressed in Kupffer cells. High levels of TGR5 are also detected in gallbladder epithelial cells, gallbladder smooth muscle cells and sinusoidal endothelial cells [27]. Thus the two BA receptors constitute a potent defense mechanism to suppress NF-κB-dependent inflammation and prevent BA-induced malignancy such as HCC (Fig. 1). As such, manipulation of BA signaling pathways mediated by FXR and TGR5 could be considered as novel drug targets for the prevention and treatments of liver cancer.

Activation of FXR and TGR5 suppress proinflammatory cytokine expression in the liver. Bile acids (BAs) activate FXR in hepatocytes to suppress cytokine expression and BA production. BAs also activate TGR5 in Kupffer cells to suppress cytokine expression and inflammation. However, the cross talk between FXR and TGR5 is unclear
References
Scheimann AO, Strautnieks SS, Knisely AS, Byrne JA, Thompson RJ, et al (2007) Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr 150:556–559
Cameron RG, Imaida K, Tsuda H, Ito N (1982) Promotive effects of steroids and bile acids on hepatocarcinogenesis initiated by diethylnitrosamine. Cancer Res 42:2426–2428
Tsuda H, Masui T, Imaida K, Fukushima S, Ito N (1984) Promotive effect of primary and secondary bile acids on the induction of gamma-glutamyl transpeptidase-positive liver cell foci as a possible endogenous factor for hepatocarcinogenesis in rats. Gann 75:871–875
Kitazawa S (1993) Studies on initiating activity of secondary bile acids for rat hepatocarcinogenesis. Hokkaido Igaku Zasshi 68:110–120
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, et al (2004) NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431:461–466
•• Yang F, Huang X, Yi T, Yen Y, Moore DD, et al (2007) Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 67:863–867. The first evidence that, in the absence of FXR, HCC develops spontaneously in mice.
Yerushalmi B, Dahl R, Devereaux MW, Gumpricht E, Sokol RJ (2001) Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 33:616–626
Gupta S, Natarajan R, Payne SG, Studer EJ, Spiegel S, et al (2004) Deoxycholic acid activates the c-Jun N-terminal kinase pathway via FAS receptor activation in primary hepatocytes. Role of acidic sphingomyelinase-mediated ceramide generation in FAS receptor activation. J Biol Chem 279:5821–5828
Jean-Louis S, Akare S, Ali MA, Mash EA, Jr., Meuillet E, et al (2006) Deoxycholic acid induces intracellular signaling through membrane perturbations. J Biol Chem 281:14948–14960
Dent P, Fang Y, Gupta S, Studer E, Mitchell C, et al (2005) Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology 42:1291–1299
Wang YD, Chen WD, Huang W (2008) FXR, a target for different diseases. Histol Histopathol 23:621–627
Chen WD, Wang YD, Meng Z, Zhang L, Huang W (2011) Nuclear bile acid receptor FXR in the hepatic regeneration. Biochim Biophys Acta 1812:888–892
•• Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, et al (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298:714–719. The first publication to identify TGR5 as a BA membrane receptor.
Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, et al (2003) A G protein-coupled receptor responsive to bile acids. J Biol Chem 278:9435–9440
• Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, et al (2006) Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol 191:197–205. TGR5 knockout mice were generated.
• Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, et al (2006) Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J 398:423–430. TGR5 knockout mice and cholestreol gallstone formation.
Chen X, Lou G, Meng Z, Huang W (2011) TGR5: a novel target for weight maintenance and glucose metabolism. Exp Diabetes Res 2011:853501
Fiorucci S, Mencarelli A, Palladino G, Cipriani S (2009) Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol Sci 30:570–580
Keitel V, Häussinger D (2012) Perspective: TGR5 (Gpbar-1) in liver physiology and disease. Clin Res Hepatol Gastroenterol 36:412–419
•• Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, et al (1995) Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 81:687–693.First paper to clone FXR gene.
Cai SY, Xiong L, Wray CG, Ballatori N, Boyer JL (2007) The farnesoid X receptor FXRalpha/NR1H4 acquired ligand specificity for bile salts late in vertebrate evolution. Am J Physiol Regul Integr Comp Physiol 293:R1400–1409
Meng Z, Wang Y, Wang L, Jin W, Liu N, et al (2010) FXR regulates liver repair after CCl4-induced toxic injury. Mol Endocrinol 24:886–897
Meng Z, Liu N, Fu X, Wang X, Wang YD, et al (2011) Insufficient bile acid signaling impairs liver repair in CYP27(−/−) mice. J Hepatol 55:885–895
• Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, et al (2006) Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 312:233–236. Identification of an unexpected role of FXR in liver regeneration.
Zhang LS, Huang XF, Meng ZP, Dong BN, Shiah S, et al (2009) Significance and mechanism of CYP7a1 gene regulation during the acute phase of liver regeneration. Mol Endocrinol 23:137–145
• Kim I, Morimura K, Shah Y, Yang Q, Ward JM, et al (2007) Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 28:940–946. First evidence of FXR in suppressing HCC.
Zhu Y, Li F, Guo GL (2011) Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol Res 63:259–265
Zhang L, Wang YD, Chen WD, Wang X, Lou G, et al (2012) Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine. Hepatology. doi:10.1002/hep.25905. FXR couples BA regulation to liver regeneration/repair. Both liver- and intestine-FXR are activated after liver resection or injury, which contribute to promoting liver regeneration/repair.
Ueno Y, Moriyama M, Uchida T, Arakawa Y (2001) Irregular regeneration of hepatocytes is an important factor in the hepatocarcinogenesis of liver disease. Hepatology 33:357–362
Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, et al (2011) Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther 338:12–21
Zhang YQ, Ge XM, Heemstra LA, Chen WD, Xu JS, et al (2012) Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol Endocrinol 26:272–280
Schoenborn JR, Wilson CB (2007) Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol 96:41–101
Meng Z, Wang X, Gan Y, Zhang Y, Zhou H, et al (2012) Deletion of IFNγ enhances hepatocarcinogenesis in FXR knockout mice. J Hepatol 57:1004–1012
•• Wang Y-D, Chen W-D, Yu D, Forman BM, Huang W (2011) The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 54:1421–1432. TGR5 suppresses hepatic inflammation by antagonizing NF-κB activities. This may be also the mechanism by which TGR5 suppresses immune response.
•• Wang YD, Chen WD, Wang M, Yu D, Forman BM, et al (2008) Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48:1632–1643. First evidence that FXR suppress hepatic inflammatory response by antagonizing NF-κB activities.
Xu Z, Huang G, Gong W, Zhao Y, Zhou P, et al (2012) Activation of Farnesoid X Receptor Increases the Expression of Cytokine Inducible SH2-Containing Protein in HepG2 Cells. J Interferon Cytokine Res 32:517–523
Wang YD, Yang F, Chen WD, Huang X, Lai L, et al (2008) Farnesoid X receptor protects liver cells from apoptosis induced by serum deprivation in vitro and fasting in vivo. Mol Endocrinol 22:1622–1632
Nomoto M, Miyata M, Yin S, Kurata Y, Shimada M, et al (2009) Bile Acid-Induced Elevated Oxidative Stress in the Absence of Farnesoid X Receptor. Biol Pharm Bull 32:172–178
Fickert P, Fuchsbichler A, Moustafa T, Wagner M, Zollner G, et al (2009) Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol 175:2392–2405
• He N, Park K, Zhang Y, Huang J, Lu S, et al (2008) Epigenetic inhibition of nuclear receptor small heterodimer partner is associated with and regulates hepatocellular carcinoma growth. Gastroenterology 134:793–802. The role of Shp in suppressing HCC is demonstrated. This paper also examines the expression of Shp in human HCC samples.
Deuschle U, Schuler J, Schulz A, Schluter T, Kinzel O, et al (2012) FXR Controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an Orthotopic mouse xenograft model. PLoS One 7:e43044
Su H, Ma C, Liu J, Li N, Gao M, et al (2012) Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am J Physiol Gastrointest Liver Physiol. 303:G1245–G1253
Fujino T, Takeuchi A, Maruko-Ohtake A, Ohtake Y, Satoh J, et al (2012) Critical role of farnesoid X receptor (FXR) for hepatocellular carcinoma cell proliferation. J Biochem. [Epub ahead of print]
Takahara Y, Takahashi M, Zhang QW, Wagatsuma H, Mori M, et al (2008) Serial changes in expression of functionally clustered genes in progression of liver fibrosis in hepatitis C patients. World J Gastroenterol 14:2010–2022
Liu N, Meng Z, Lou G, Zhou W, Wang X, et al (2012) Hepatocarcinogenesis in FXR−/− mice mimics human HCC progression that operates through HNF1alpha regulation of FXR expression. Mol Endocrinol 26:775–785
Zhang Y, Gong W, Dai SS, Huang G, Shen XD, et al (2012) Downregulation of human Farnesoid X receptor by miR-421 promotes proliferation and migration of hepatocellular carcinoma cells. Mol Cancer Res 10:516–522
Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, et al (2007) The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology 45:695–704
Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, et al (2009) The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50:861–870
Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, et al (2005) International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev 57:279–288
Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, et al (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489
Houten SM, Watanabe M, Auwerx J (2006) Endocrine functions of bile acids. EMBO J 25:1419–1425
Thomas C, Auwerx J, Schoonjans K (2008) Bile acids and the membrane bile acid receptor TGR5–connecting nutrition and metabolism. Thyroid 18:167–174
Sato H, Genet C, Strehle A, Thomas C, Lobstein A, et al (2007) Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun 362:793–798
Katsuma S, Hirasawa A, Tsujimoto G (2005) Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun 329:386–390
• Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, et al (2009) TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10:167–177.TGR5 participates in regulating glucose homeostasis.
Trauner M, Claudel T, Fickert P, Moustafa T, Wagner M (2010) Bile acids as regulators of hepatic lipid and glucose metabolism. Dig Dis 28:220–224
Martin R, Carvalho-Tavares J, Hernandez M, Arnes M, Ruiz-Gutierrez V, et al (2010) Beneficial actions of oleanolic acid in an experimental model of multiple sclerosis: a potential therapeutic role. Biochem Pharmacol 79:198–208
Schmidt SV, Nino-Castro AC, Schultze JL (2012) Regulatory dendritic cells: there is more than just immune activation. Front Immunol 3:274
Ichikawa R, Takayama T, Yoneno K, Kamada N, Kitazume MT, et al (2012) Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 136:153–162
•• Chen W-D, Yu D, Forman BM, Huang W, Wang Y-D (2012) The deficiency of G-protein-coupled bile acid receptor gpbar1 (TGR5) enhances chemically-induced liver carcinogenesis. Hepatology. doi:10.1002/hep.26019. First evidence that TGR5 may suppress HCC development.
He G, Karin M (2011) NF-kappaB and STAT3: key players in liver inflammation and cancer. Cell Res 21:159–168
Fiorucci S, Mencarelli A, Distrutti E, Zampella A (2012) Farnesoid X receptor: from medicinal chemistry to clinical applications. Future Med Chem 4:877–891
Hollman DAA, Milona A, van Erpecum KJ, van Mil SWC (2012) Anti-inflammatory and metabolic actions of FXR: Insights into molecular mechanisms. Biochimica Biophysica Acta 1821:1443–1452
Fu J, Si P, Zheng M, Chen L, Shen X, et al (2012) Discovery of new non-steroidal FXR ligands via a virtual screening workflow based on Phase shape and induced fit docking. Bioorg Med Chem Lett 22:6848–6853
Fiorucci S, Distrutti E, Bifulco G, D’Auria MV, Zampella A (2012) Marine sponge steroids as nuclear receptor ligands. Trends Pharmacol Sci 33:591–601
Ono E, Inoue J, Hashidume T, Shimizu M, Sato R (2011) Anti-obesity and anti-hyperglycemic effects of the dietary citrus limonoid nomilin in mice fed a high-fat diet. Biochem Biophys Res Commun 410:677–681
Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, et al (2011) The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PloS One 6:e25637
Papers of particular interest have been highlighted as: • Of importance •• Of major importance
Jenkins G, Hardie LJ, Royal Society of Chemistry (2008) Bile acids : toxicology and bioactivity. Royal Society of Chemistry, Cambridge
Halilbasic E, Claudel T, Trauner M (2012) Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. doi:10.1016/j.jhep.2012.08.002
Liao M, Zhao J, Wang T, Duan J, Zhang Y, et al (2011) Role of bile salt in regulating Mcl-1 phosphorylation and chemoresistance in hepatocellular carcinoma cells. Mol Cancer 10:44
Tinkle CL, Haas-Kogan D (2012) Hepatocellular carcinoma: natural history, current management, and emerging tools. Biologics 6:207–219
Baptissart M, Vega A, Maqdasy S, Caira F, Baron S, et al (2012) Bile acids: from digestion to cancers. Biochimie. [Epub ahead of print]
Raufman JP, Cheng K, Zimniak P (2003) Activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig Dis Sci 48:1431–1444
Jansen PLM (2007) Endogenous bile acids as carcinogens. J Hepatol 47:434–435
•• Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, et al (2006) Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology 44:478–486. The first indication of a link between BAs and human HCC.
Acknowledgments
We apologize to those colleagues whose work could not be cited due to space limitations. We thank Drs. David Moore, Barry Forman, Donna Yu, and Yan-Dong Wang for their contribution and discussion. This work is supported by NCI R01-CA139158.
Conflict of interest
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, X., Fu, X., Van Ness, C. et al. Bile Acid Receptors and Liver Cancer. Curr Pathobiol Rep 1, 29–35 (2013). https://doi.org/10.1007/s40139-012-0003-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-012-0003-6