
Abstract
We investigated the complexation ratio of siRNA and plasmid DNA combined with Lipofectamine for the effective delivery of genetic materials. First, the amount of Lipofectamine was varied with a fixed amount of DNA or siRNA to determine in which proportions they would form an optimal combination. Finally, to investigate the effect of DNA or siRNA on the co-complexation of both DNA and siRNA, the co-complex of DNA and siRNA was prepared at the various ratios with a fixed amount of DNA. All complexation was confirmed by gel retardation of DNA or siRNA on agarose gels. The effects of siRNA complexes on mRNA expression from plasmid DNA were explored post-transfection, while the influence of plasmid DNA complexes on the transfection of siRNA was determined in GFP-expressing H4IIE cells. The complex between DNA and Lipofectamine was formed at a weight ratio of 0.8:1, whereas the light band of siRNA/Lipofectamine disappeared at a weight ratio of 4:1. When the amounts of DNA, siRNA, and the mixture were fixed, the optimal ratio of nucleic acids and Lipofectamine in our composition was 110:80:350 (ng). Confocal images and flow cytometry showed that inhibition of GFP expression by siRNA was not interfered with by co-complexed plasmid DNA. Moreover, mRNA expression of adiponectin was not hampered by the addition of siRNA; rather, it was increased. Thus, co-complexation of siRNA and plasmid DNA may have a synergistic effect on delivery of the therapeutic gene and siRNA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In patients with type 2 diabetes mellitus (T2DM), plasma levels of adiponectin (ADN) are decreased significantly (Hotta et al. 2000). ADN, an adipokine hormone secreted primarily from adipose tissue, stimulates glucose uptake and fatty acid oxidation and inhibits gluconeogenesis, resulting in improved insulin sensitivity (Kola et al. 2006). ADN decreases insulin resistance by lowering the triglyceride content in muscles and the liver in obese mice via the increased expression of molecules involved in both fatty acid combustion and energy dissipation in muscles (Yamauchi et al. 2001). ADN also stimulates glucose utilization and fatty-acid oxidation by activating 5′-AMP-activated protein kinase (AMPK) (Yamauchi et al. 2002).
In contrast, the normal physiological role of resistin remains unclear, but the hormone has been linked to the development of obesity and T2DM. Resistin levels are increased in obese mice, relative to normal-weight animals, and administration of exogenous resistin to normal mice results in both impaired glucose tolerance and insulin resistance. Conversely, neutralization of resistin with an anti-resistin antibody increases insulin sensitivity and blood glucose levels in mice on a high-fat diet. Moreover, resistin levels are modulated in vitro by antidiabetic thiazolidinedione drugs (Steppan et al. 2001). To establish a therapeutic strategy for T2DM, it is necessary to elevate ADN levels and downregulate resistin levels in plasma.
Combinations of two or more therapeutic agents with pharmacodynamically synergistic or additive effects are effective against a number of cancers and various other diseases. Although drug/gene combinations have been investigated and used in the treatment of cancer, only a few reports of the simultaneous delivery of DNA and siRNA have been published to date (Kang and Bae 2011; Jeon et al. 2012).
The combination of plasmid DNA and siRNA is feasible for achieving synergistic or additive effects. Both plasmid DNA and siRNA have anionic phosphodiester backbones with identical negative charge: nucleotide ratios and should therefore interact electrostatically with cationic liposome/lipid systems to form cationic liposome/lipid-nucleic acid (“lipoplex”) particles that can transfer the nucleic acids into cells. However, plasmid DNA and siRNA are otherwise very different from each other in molecular weight and molecular topography, with potentially important consequences. The complexation ratios of plasmid DNA and siRNA to cationic liposomes should be examined to optimize transfection efficiency.
In this study, we constructed pCAG/ADN and prepared lipoplexes to deliver plasmid DNA and siRNA simultaneously. Then, we investigated the optimized complexation ratio through gel retardation and transfection, and the effects of plasmid DNA or siRNA on transfection after co-delivery.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin–streptomycin, and trypsin–EDTA were purchased from Gibco BRL (Grand Island, NY, USA). Lipofectamine 2000 and Trizol were from Invitrogen (Carlsbad, CA, USA). Distilled and deionized water was used after sterilization. All other chemicals were of reagent grade and were used without further purification.
The siRNA for the targeted silencing of GFP was purchased from Bioneer Co. (Daejeon, Korea). The sequence of the siRNA was 5′-GCA UCA AGG UGA ACU UCA A-3′ (sense), and 5′-UUG AAG UUC ACC UUG AUG C-3′ (antisense).
Construction of plasmid DNA
An ADN expression-enhanced plasmid DNA was constructed by deleting the CMV promoter from pVAX/ADN and inserting the chicken β-actin (CAG) promoter and CMV enhancer into the pVAX/ADN vector using the MluI and BamHI restriction enzymes (NEB, UK); this construct was named pCAG/ADN (Fig. 1). Plasmid DNA was prepared using the Endo-Free Qiagen kit (Qiagen, CA, USA) to remove bacterial endotoxins. The cloned plasmid DNA, pCAG/ADN was sequenced to confirm its fidelity and was compared with pVAX/ADN for the RT-PCR.

a Construction of pCAG/ADN, b in vitro transfection of pVAX/ADN and pCAG/ADN
Cell lines
Human hepatocellular carcinoma cells (HepG2) were cultured in DMEM supplemented with 10 % heat-inactivated FBS, 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C in humidified incubator in 5 % CO2. Rat hepatoma cells (H4II-E), stably expressing green fluorescent protein (GFP), were provided by Dr. SK Kim (Chungnam National University, Daejeon, Republic of Korea). The cells were cultured in DMEM in 100 mm Petri dishes in a humidified incubator in 5 % CO2 and maintained at 37 °C for 24 h. All media were supplemented with 10 % heat-inactivated fetal bovine serum, 100 U/mL of penicillin (Gibco BRL), and 100 μg/mL of streptomycin (Gibco BRL).
Preparation of DNA or siRNA complexes with transfection reagent
Lipoplexes were prepared by mixing the DNA or siRNA with Lipofectamine (Invitrogen) at various ratios according to the manufacturer’s instructions. The mixtures were incubated for 15 min at 37 °C to facilitate complex formation. The formation of these complexes was confirmed by a gel retardation assay.
Agarose gel retardation
Complex formation between the plasmid DNA and Lipofectamine was assessed using agarose gel electrophoresis. Various ratios (0.05–2, w/w) of Lipofectamine/DNA mixtures combined with a fixed amount of DNA (1 μg of pCAG/ADN) were incubated for 15 min to facilitate complex formulation and loaded onto a 1 % agarose gel. Various ratios (0.2–10, w/v) of Lipofectamine/siRNA mixtures with a fixed amount of siRNA (10 pmol siRNA) were incubated for 15 min to facilitate complex formulation and loaded onto a 2 % agarose gel. Gel electrophoresis was conducted in TBE buffer (45 mM Tris, boric acid, 1 mM EDTA) at 50 mV. The ethidium bromide-stained DNA was examined using a UV illuminator.
In vitro transfection
Hepatoma cells were seeded into six-well plates at a density of 5 × 105/well in 2 mL complete medium. After overnight incubation, the media were removed and replaced with 500 μL serum-free media and Lipofectamine complexes equivalent to 1.1 μg plasmid DNA and 30 60 pmol siRNA at the desired N/P ratios were added to each well. After 4 h, the media were replaced with 2 mL fresh medium containing 10 % FBS, and incubated for another 24 h at 37 °C (Zhang et al. 2008). All transfection experiments were performed in triplicate.
Confocal laser scanning microscopy
To visualize GFP expression in cells, confocal laser scanning microscopy was used. H4II-E cells were grown on 22 × 22 mm coverslips. The cells were transfected with Lipofectamine complexes prepared in basal DMEM. Control cells were untreated and maintained in basal DMEM. At 24 h after transfection, the cells were rinsed with PBS and observed with confocal microscopy (Leica TCS NT, Leica Microsystems, Wetzlar, Germany) equipped with a diode laser and associated filters for simultaneous 488 nm excitation.
Flow cytometry
For comparison of transfection efficiency, mean GFP intensity was measured by flow cytometry. The cells were transfected with Lipofectamine/siRNA complexes prepared in basal DMEM and incubated for 24 h. After medium was removed and the cells were washed twice with pre-cold PBS, cells were transferred into a 5 mL polystyrene round-bottom tube (BD Falcon, Bedford, MA, USA) using 200 μL pre-cooled fresh medium and maintained at 4 °C for later flow cytometry analysis. GFP mean intensity of the H4II-E cells was recorded in the FL1 channel using a FACSCalibur (Becton–Dickinson, USA) flow cytometer (Zou et al. 2005).
Reverse transcriptase-polymerase chain reaction analysis
The transfected cells were lysed with Trizol (Invitrogen) and protein was extracted with chloroform. Total RNAs were precipitated with isopropanol and washed twice with 70 % ethanol, diluted with DEPC-treated water. The purity and concentration of RNAs were analyzed using a spectrophotometer at 260 and 280 nm. cDNA was synthesized from total RNA in a reaction mixture containing 5× reaction buffer (250 mM Tris–HCl pH 8.3, 375 mM KCl, 15 mM MgCl2, 50 mM DTT), 100 μg/mL oligo(dT), 10 mM dNTP, 100 U M-MLV reverse transcriptase (Promega, Madison, WI). The RT reaction product was amplified by PCR with forward 5′-GGA ATT CAT GCT ACT GTT GCA AGC TCT-3′ and reverse 5′-GCT CTA GAT CAG TTG GTA TCA TGG TAG AGA AG-3′ for mouse ADN, and with forward 5′-ATC CCA TCA CCA TCT TCC AG-3′ and reverse 5′-ACC TGC TTA CAC CAC CTT G-3′ for human GAPDH as an example house-keeping gene. Amplification of mouse ADN was performed with 30 cycles at 94 °C for 30 s, 59 °C for 40 s and 72 °C for 1 min. PCR products were separated on a 1 % agarose gel and visualized by UV. cDNA was quantified using the ChemiDoc XRS System (Quantity One, Bio-Rad) to quantitate ADN mRNA or GAPDH mRNA in separate reactions.
Statistical analysis
Statistical analysis of the data was performed using student’s t test or one-way analysis of variance (ANOVA). A p value of <0.05 was considered to indicate statistical significance. All data are expressed as mean ± SD from three independent experiments.
Results and discussion
In vitro transfection
To explore the feasibility of pCAG/ADN, transfection of ADN-encoding plasmid DNA was investigated in HepG2 cells. The ratio of lipoplexes was fixed as 1:2.5 (w/w) and the dose of DNA was increased, and vice versa. The elevation of ADN mRNA expression levels by gene transfer was dose-dependent with the two plasmids, pCAG/ADN and pVAX/ADN (Fig. 1b). The ADN mRNA expression levels with pCAG/ADN were twofold higher than those with pVAX/ADN, which increased dose-dependently in most cell lines transfected (Nan et al. 2010). Thus, it is thought that the newly constructed plasmid DNA (pCAG/ADN) was an effective approach to improving the expression of ADN in the target organ, the liver.
Gel retardation of DNA or siRNA lipoplexes
The nucleic acids complexed with Lipofectamine, were studied using gel retardation assays according to a previous report (Jeong et al. 2009). To verify Lipofectamine/DNA complex formation, agarose gel electrophoresis was performed after the complexes were formed with 100 ng/μL pCAG/ADN. Free DNA that failed to complex with Lipofectamine was clearly visible at complex ratios of 0.05–2:1 (μg:μg). However, free DNA disappeared when the ratio of Lipofectamine to DNA was greater than 0.83:1 (μg:μg) (Fig. 2a). The movement of plasmid DNA on a 1 % agarose gel was retarded as the amount of Lipofectamine increased. At ratios above 0.8, plasmid DNA was able to bind to Lipofectamine effectively, and the complete retardation of DNA mobility was achieved at a 1:1 ratio. Complexes at ratios below 0.67 formed incomplete complexes.

Gel retardation assay of lipoplexes at various ratios. a Lipofectamine/DNA ratio (w/w), b Lipofectamine/siRNA ratio (w/w), c Lipofectamine/siRNA (40 ng)/DNA (110 ng), d Lipofectamine (350 ng)/siRNA/DNA (2 % agarose gel), e Lipofectamine (350 ng)/siRNA/DNA (1 % agarose gel)
To confirm complex formation between Lipofectamine and the negatively charged siRNA, we used an agarose gel retardation assay. To verify Lipofectamine/siRNA complex formation, agarose gel electrophoresis was performed after the complexes were formed with 10 pmol/μL (80 ng) of siRNA. Free siRNA that failed to complex with Lipofectamine was clearly visible at complex ratios of 0.2–10:1 (μg:μg). When the complexation weight ratios of Lipofectamine:siRNA were less than 4:1, no retardation of siRNA was observed. However, gel retardation was clearly indicated when the complexation weight ratio of Lipofectamine:siRNA was greater than 4:1 (Fig. 2b).
To identify the optimal proportions, co-complexes of DNA and siRNA were prepared at various ratios with a fixed amount of DNA and siRNA, 110 and 40 ng, respectively. Then, the amount of Lipofectamine was altered at various ratios and the formation of complexation was confirmed by gel retardation in a 2 % agarose gel. From these results, the amount of Lipofectamine was fixed at 330 ng (Fig. 2c). To investigate whether a co-complex of DNA and siRNA was formed, the amount of siRNA was varied from 40 to 160 ng with the amount of DNA fixed at 110 ng, and the amount of DNA was increased from 150 to 300 ng at a fixed amount of 40 ng siRNA. Although it was found that all DNA was complexed with Lipofectamine, the siRNA band began to disappear at 120 ng DNA. Thus, the minimum amounts of DNA and siRNA were 110 and 40 ng, respectively. As shown in Fig. 2d and e, siRNA and plasmid DNA were retarded in the agarose gels (2 and 1 %, respectively). With fixed amounts of DNA (110 ng) and siRNA (40 ng), the appropriate complexation mixture was 330 ng Lipofectamine.
Effect of plasmid DNA on GFP inhibition by siRNA
In a previous study, the optimum complexation ratio of DNA, siRNA and Lipofectamine was 110:80:350 (ng), Based on this ratio, various compositions of transfection complexes were applied to H4II-E cells. Silencing of GFP expression by siRNA following the vitro transfection experiment was observed using a confocal microscope at 24 h after the Lipofectamine-mediated transfection of the H4II-E cells. GFP expression was evident in intact H4II-E control cells (Fig. 3a), whereas the GFP intensity in the cells treated with complexed siRNA was reduced, with slight differences depending on formulation, as in the micrographs (Fig. 3c–f). Naked siRNA showed higher GFP intensity than any other treated groups, showing the low transfection efficiency of siRNA alone (Fig. 3b).

Inhibition of GFP-expressing H4ll-E cells after treatment with Lipofectamine with GFP-siRNA and DNA (n = 3). a Untreated H4II-E cells, b treated with siRNA alone, c treated with Lipofectamine/DNA complex, d treated with Lipofectamine/siRNA complex, e treated with Lipofectamine/siRNA (40 ng)/DNA complex, and f treated with Lipofectamine/siRNA (80 ng)/DNA complex
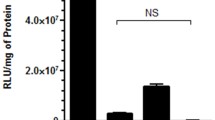
As shown in Fig. 4, complexation of Lipofectamine increased the transfection efficiency of siRNA alone by approximately 12 %. However, the silencing effect of siRNA was not affected by co-complexed plasmid DNA. Moreover, increased amounts of siRNA, from 40 to 80 ng, showed significant decreases. It was supposed that the transfection efficiency of triple combination lipoplexes would be related to the amount of siRNA. Thus, the complexation ratio of DNA, siRNA, and Lipofectamine (110:80:350) could increase the transfection efficiency of siRNA to 20 %. Our results are consistent with a study that did not include a specific delivery system for application of siRNA (Sørensen et al. 2003).

Inhibition of GFP expression in H4ll-E cells after treatment with Lipofectamine with GFP-siRNA and DNA (n = 3)
Effect of siRNA on mRNA expression from DNA
To examine the influence of siRNA on the transfection of plasmid DNA, the amount of cationic lipid was fixed at 350 ng and the complexation ratio of DNA and siRNA was varied. As shown in Fig. 5, the increase in mRNA expression of ADN was dependent on the lipoplex ratio. Moreover, mRNA expression of ADN was increased by addition of siRNA, rather than inhibited by it. Plasmid DNA and siRNA could be formulated differently with cationic liposomes and lipids, and mechanically because of their sizes, the two species of nucleic acids vary substantially, both in terms of intracellular uptake and accumulation into defined intracellular vesicles (Spagnou et al. 2004). Because the mechanism underlying the enhancement of mRNA expression from DNA by siRNA remains unknown, further studies are needed.

ADN mRNA expression in H4ll-E cells after treatment with Lipofectamine with GFP-siRNA and plasmid DNA (n = 3)
Conclusions
When the amounts of the DNA, siRNA, and mixture were fixed, the optimized ratio of nucleic acids and Lipofectamine in our composition was found to be 110:80:350 (ng). To some extent, the inhibition of GFP expression by siRNA was interfered with by the co-complexed plasmid DNA. However, mRNA expression of ADN was not hampered by addition of siRNA; rather, it was increased. Thus, co-complexation of siRNA and plasmid DNA may potentially have synergistic effects with therapeutic genes and siRNA.
References
Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, Iwahashi H, Kuriyama H, Ouchi N, Maeda K, Nishida M, Kihara S, Sakai N, Nakajima T, Hasegawa K, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Hanafusa T, Matsuzawa Y (2000) Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 20(6):1595–1599
Jeon SY, Park JS, Yang HN, Woo DG, Park KH (2012) Co-delivery of SOX9 genes and anti-Cbfa-1 siRNA coated onto PLGA nanoparticles for chondrogenesis of human MSCs. Biomaterials 33(17):4413–4423
Jeong UH, Jung JH, Davaa E, Park SJ, Myung CS, Park JS (2009) Effect of drug loading on the physicochemical properties and stability of cationic lipid-based plasmid DNA complexes. J Korean Pharm Sci 39(5):339–343
Kang HC, Bae YH (2011) Co-delivery of small interfering RNA and plasmid DNA using a polymeric vector incorporating endosomolytic oligomeric sulfonamide. Biomaterials 32(21):4914–4924
Kola B, Boscaro M, Rutter GA, Grossman AB, Korbonits M (2006) Expanding role of AMPK in endocrinology. Trends Endocrinol Metab 17(5):205–215
Nan MH, Park JS, Myung CS (2010) Construction of adiponectin-encoding plasmid DNA and gene therapy of non-obese type 2 diabetes mellitus. J Drug Target 18(1):67–77
Sørensen DR, Leirdal M, Sioud M (2003) Gene silencing by systemic delivery of synthetic siRNAs in adult mice. J Mol Biol 327(4):761–766
Spagnou S, Miller AD, Keller M (2004) Lipidic carriers of siRNA: differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry 43(42):13348–13356
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA (2001) The hormone resistin links obesity to diabetes. Nature 409(6818):307–312
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T (2001) The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7(8):941–946
Yamauchi T, Kamon J, Miniokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8(11):1288–1295
Zhang Z, Sha X, Shen A, Wang Y, Sun Z, Gu Z, Fang X (2008) Polycation nanostructured lipid carrier, a novel nonviral vector constructed with triolein for efficient gene delivery. Biochim Biophys Res Commun 370(3):478–482
Zou C, Wang Y, Shen Z (2005) 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J Biochem Biophys Methods 64(3):207–215
Acknowledgments
This article does not contain any studies with human and animal subjects performed by any of the authors. And all authors (E Davaa, IH Ahn, BS Kang, SE Lee, CS Myung and JS Park) declare that they have no conflict of interest. This research was supported by the Basic Science Research Program (No. 2010-0003083) and the Priority Research Centers Program (No. 2012-0006681) through the National Research Foundation of Korea (NRF), funded by the Ministry of Education.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davaa, E., Ahn, IH., Kang, BS. et al. Preliminary study to determine the optimal conditions for the simultaneous complexation of siRNA and plasmid DNA. Journal of Pharmaceutical Investigation 43, 499–505 (2013). https://doi.org/10.1007/s40005-013-0095-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40005-013-0095-y