
Abstract
The interest in hot-melt extrusion as a drug delivery technology for the production of solid dispersion is growing rapidly. Lumefantrine (LUMF) is an antimalarial drug that exhibits poor oral bioavailability, in consequence of its poor aqueous solubility. To improve its antimalarial activity, solid dispersion formulation using hot melt extrusion technology was prepared. Appropriate selection of polymers, favoured the production of amorphous LUMF-polymer solid dispersions. The physicochemical properties of solid dispersions were characterized using scanning electron microscope, Infrared spectroscopy, differential scanning calorimetry and X-ray diffraction. LUMF SD showed enhanced dissolution rate attributed to amorphosization of LUMF. The IC50 value of LUMF SD formulations was found to be (0.084–0.213 ng/mL) i.e. 220–101 times lower than the IC50 value of pure LUMF (18.2 ng/mL) and 45–18 times lower than the IC50 value of standard antimalarial drug, chloroquine (3.8 ng/mL). Molecular dynamic simulation approach was used to investigate drug-polymer molecular interaction using computational modelling Schrodinger® software. LUMF SD powder makes the Coartem® therapy more operative with value-added beneficial comeback.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Although having an abundant therapeutic activity, many drug substances under development exhibit low aqueous solubility and consequently, poor bioavailability. Low aqueous solubility can chiefly be ascribed to high intermolecular forces contained by the crystal lattice, high lipophilicity or a grouping of these elements. Taming the aqueous solubility of these compounds, even momentarily, can expressively impact their absorption in the GI tract when medicated orally. To this end, enhancing the solubility of this class of drug substances has become a foremost topic of importance in the drug industry. Oral drug administration is the most suitable medication treatment and repay more than 60 % stake of the marketed drug formulations. In recent years there has been a unswerving trend in drug discovery towards identification of poorly soluble molecules as lead candidates for oral administration and at present about 40 % of the drugs in the development pipelines are poorly soluble (Juma 2008; Andriantsoanirina 2011). Formulation development of such compounds is thought-provoking and problems related to dissolution and bioavailability of such compounds is perilous and may necessitate the use of novel technologies (Chiou and Riegelman 1971). Various technologies to augment the dissolution rate and bioavailability of these candidates, together with use of salt formation, micronization, pH-modification, emulsification, micellar dispersions and complexation with cyclodextrin have established substantial interest (Ford 1986). An alternative to such methods is hot melt extrusion technique. HME leads to an enhanced dissolution rate not only because of increased surface area, but also because of increased saturation solubility as described by Gordon-Taylor equation (Ambike et al. 2004). HME is cost effective, fast, solvent free and easy to scale up technique. In addition, some drugs are degraded by the thermal energy generated by HME (Doshi and Betageri 1997). The glass transition temperature (Tg) of polymers and that of drug, melting point of drug are the essential factors responsible for the intermixing of drug–polymer at molecular level. HME not only has the ability to convert crystalline form of drug into amorphous but also drug remains in amorphous stable form. The homogenously dispersed materials are stable in nature (Takeuchi and Nagira 2004). Several techniques for controlled, immediate, sustained release formulation using HME have been reported.
Based on these considerations, the objective of the present investigation was to formulate HME processed solid dispersion powder of poorly water soluble drug Lumefantrine (LUMF) and to evaluate it for the antimalarial efficacy. LUMF is a potent antimalarial agent available in combination with artemether in a 6:1 ratio (Coartem®) for the treatment of severe multiresistant malaria (Suneela 2005). The combination is active against Plasmodium vivax as well as against chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum and is also indicated in the treatment of cerebral malaria (Crowley et al. 2002). However, the therapeutic potential of drug is significantly hindered due to its low and inconstant oral bioavailability (Repka et al. 2003). The low and inconstant oral bioavailability of LUMF curtails from its poor aqueous solubility. In construal to this, the SD of LUMF is likely to have great potential in improving oral bioavailability, solubility, dissolution rate and in turn the therapeutic efficacy of LUMF. Improving the aqueous solubility of LUMF can significantly impact their absorption in the GI when delivered orally (Hancock and Zografi 1997). To this end, enhancing the solubility of this class of drug substances has become a major point of interest in the pharmaceutical industry. The main objective of the investigation was to formulate and evaluate the efficacy of a commercially feasible oral delivery strategy for LUMF.
Hot melt extruded solid dispersion systems have been explored extensively over the last several decades with a number of advancements being made (Zhang and McGinity 1999). It has become apparent that polymers with both hydrophilic and hydrophobic functional groups are well-suited for use as carriers in which the drug substance is dissolved at the molecular level (McGinity et al. 2001). Interactions between polymer and drug functional groups are capable of not only kinetically stabilizing the amorphous form in the solid-state, but also inhibiting precipitation in the liquid state, effectively providing maintenance of supersaturation (Vasconcelos et al. 2007). A number of mechanisms have been proposed to describe the method by which the precipitation is inhibited by an additive such as a polymer. In reflection to current study, these include alteration of surface tension or saturation solubility, production of intermolecular bonding charges at crystal-medium interface and absorption onto a crystal interface consequently hindering further crystallization (Chokshi et al. 2007). The use of cellulose-based polymeric carriers in solid dispersion systems is becoming relatively common due to their effectiveness as precipitation inhibitors.
In the present study, we are investigating the effect of polymers Soluplus, Kollidon VA64 and Plasdone S630 in terms of solubilisation and dissolution rate enhancement of LUMF using hot melt extrusion technology. However there is no literature reported on enhancement of solubility and dissolution rate of LUMF by hot melt extrusion method.
Materials and methods
Materials
Lumefantrine was obtained as a gift sample from Bajaj Healthcare Pvt. Ltd. (India). Soluplus, Kollidone VA64 were generously gifted from BASF AG (Germany). Plasdone S630 was gift from ISP polymers, India. Water was produced in the laboratory by a Milli-Q purification system (Millipore, Billerica, MA, USA). All the other chemicals used were of analytical grade (Fig. 1).

Chemical structure of lumefantrine
Method of preparation of hot-melt extrudates
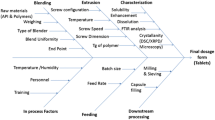
Solid dispersions (SD) were prepared by hot melt extrusion in a single-screw extruder (Manufactured by S.B. Panchal Ltd. Mumbai, India). Extrusion parameters were adjusted for drug and polymer are summarized in Table 1. Die used for extrusion was of 2 mm diameter. LUMF was mixed with Soluplus, Kollidone VA64 and Plasdone S630 at drug/polymer mass ratios of 1:1, 1:2 and 1:3 using a mortar and pestle for 5 min. The prepared physical mixtures (PMs) were extruded using a corotating single-screw extruder at a screw speed of 50 rpm. The temperatures for processing were selected based on the Tg of the polymers and melting point of the drug. As a general rule, an extrusion process should be conducted at temperatures 20–40 °C above the Tg of the polymer and at a temperature close to the melting point of the drug. The temperatures employed were 115, 110 and 113 °C for Soluplus, Kollidone VA64 and Plasdone S-630 systems, respectively. Drug content analyses and stability studies were carried out by using developed HPLC method. While other parameters like solubility, phase solubility, dissolution rate were measured with the help of UV-spectroscopy.
Solubility parameter calculations
Solubility parameter (δ) for LUMF was performed by the group contribution method using molecular modelling pro software (Schrodinger Maestro, USA). The solubility parameters for the polymers were taken from the literature and matched to the LUMF by observing the relative difference in total, ∆δ (Greenhalgh and Timmins 1999).
Density measurements
The true density of the LUMF and polymers were determined in duplicate using a gas displacement pycnometer (Accupyc 1330; Micromeritics, Norcross, Georgia) (Konno and Taylor 2006).
Drug content
The assay of the melt extrudates was assessed using high-performance liquid chromatography (HPLC) apparatus equipped with a quipped with Binary HPLC pump, and 2998 UV array detector (Agilent Corporation, Milford, Massachusetts). A reverse-phase C18 column (150 × 4.6 mm; 5 μm particles) was used. The mobile phase acetonitrile—0.1 M ammonium acetate buffer adjusted to pH 4.9 (85:15 %, v/v) was used as the mobile phases using same apparatus as in case of LUMF (Gahoia and Jain 2012). Samples equivalent to 120 mg of LUMF were dissolved in 5 mL of methanol and appropriately diluted and the drug content was determined by HPLC at λ = 338 nm.
Saturation solubility
An excess amount of the SD formulation was added to 10 mL of the 0.1 N HCl with 1 % Benzalkonium chloride (BKC), in water and sonicated at ambient room temperature (∼37 °C) for 25 min (Makar et al. 2013). All test tubes were covered with aluminium foil and subjected to sonication for 20 min at room temperature. Then were kept in orbital shaking thermo stable incubator (Boekel Scientific, Germany) for 48 h at 37 ± 0.5 °C with rotating speed of 75 agitations/min. The supernatant solution was then passed through a Whatman filter paper (Grade 1). After equilibration, the samples were filtered through 0.45 μm pore size nylon filters suitably diluted, and analysed by UV at 338 nm. Along with pure LUMF solubility of prepared SD was also carried out in these media. All solubility measurements were performed in triplicate.
Phase solubility study
Phase solubility study was performed according to the method described by Higuchi and Connors (Higuchi and Connors 1965). An excess amount of LUMF was placed in 20 mL test tubes containing in 10 mL of distilled water with different concentrations of Soluplus, Kollidone VA64 and Plasdone S630 separately. Soluplus (1, 2, 3, 4 and 5 % w/v) for was used as hydrophilic polymer and same % used for Kollidon VA64, Plasdone S630 in different sets of test tubes. Test tubes were covered with cellophane membrane to avoid solution loss and then shaken (75 agitations/min) in orbital shaking incubator (Boekel Scientific, Germany) for 48 h at 37 °C. The solutions in the test tubes were vortexed and kept for centrifugation for 20 min at 10,000 rpm. 5 mL of supernatant was withdrawn and filtered through Whatmann filter paper (Grade 1). The filtrates were analysed using a UV–Vis spectrophotometer at 338 nm after suitable dilution. All solubility measurements were performed in triplicate.
ΔG°tr-Gibbs-free energy calculation
The ΔG°tr value provides information about whether the treatment is favourable or unfavourable for drug solubilization in an aqueous medium. Negative Gibbs-free energy values indicate improved dissolution (Zsombor and Attila 2012). The ΔG°tr values of LUMF were calculated using the following equation:
where S0/Ss, is the ratio of the molar solubility of LUMF before and after treatment with mixture of polymer Soluplus, Kollidone VA64 and Plasdone S630. The value of gas constant (R) is 8.31 J/K mol and T is temperature in degree kelvin. The order of Phase solubility and ΔG°tr of LUMF at different concentrations of Soluplus, Kollidone VA64 and Plasdone S630 shown.
Flory–Huggins modelling
The Flory–Huggins (FH) interaction parameter (χ) was estimated from melting point depression data. The FH interaction parameter (χ) was calculated using the following equation:
where Tm mix is the melting temperature of the drug in the presence of the polymer, Tm pure is the melting temperature of the drug in the absence of the polymer, ∆H f is the heat of fusion of the pure drug, m is the ratio of the volume of the polymer to that of LUMF, and Φ drug and Φ polymer are the volume fractions of the drug and the polymer, respectively (Linn 2012).
Gordon-Taylor theory
Thermal analysis by DSC drug–polymer miscibility is the key factor for the stability of amorphous pharmaceutical solid dispersion systems; partial miscibility or poor solubility can result in the formation of concentrated drug domains that may be prone to recrystallization after production and during storage. Miscibility of the drug with the polymer can be assessed based upon the shift in melting endotherm or Tg of the drug or can be predicted theoretically using the Gordon-Taylor equation based on the Tg, densities, and weight fractions of the pure components (Breitenbach et al. 2002).
where, Tg is the glass transition temperature, W1 and W2 are the weight fractions of the components, and K is the parameter calculated from the true densities (ρ) and Tg of the components.
Characterization of hot-melt extrudates
The melt extrudates were grounded and passed through a number 60 sieve. The formulations were analysed for drug content and saturation solubility and further characterized by differential scanning calorimetry (DSC), X-ray diffraction, SEM and Fourier transform infrared (FTIR) spectroscopy analyses.
Differential scanning calorimetry (DSC)
Differential scanning calorimeter (DSC-PYRIS-1, Perkin Elmer, USA) was used to study the drug polymer interactions and thermal behaviour of drug. The experiments were performed in a dry nitrogen atmosphere. The samples were heated at a rate of 10 °C/min from ambient temperature to the melting point. Samples (5.0–10.0 mg) of LUMF, Soluplus, Kollidone VA64, Plasdone S630 and extrudates were accurately weighed into crimped aluminium pans and heated at 10 °C/min under a nitrogen purge (20 mL/min) from 0 to 160 °C. Solid dispersion samples were cooled rapidly from 160 °C/min to 40 °C and reheated at 10 °C/min (second heating cycle) to 160 °C. In order to understand the miscibility of Soluplus, Kollidone VA64, Plasdone S630 and SDs were investigated. An empty crimped aluminium pan was used as the reference cell. The DSC was calibrated for baseline using empty cells, and for temperature.
X-ray powder diffraction (XPRD)
The crystallinity between two samples was measured using a Miniflex apparatus (Rigaku, Japan) with CuKα radiation. Samples were held on quartz frame. Diffraction pattern were obtained at a voltage of 45 kV and at a current of 20 mA. The slide was then placed vertically at 0° angle in the X-ray diffractometer so that the X-ray beam fell on it properly. The results were recorded over a range of 0–40° (2θ) using the Cu-target X-ray tube and Xe-filled detector. The operating conditions were: voltage 40 kV; current 20 mA; scanning speed 1/min; temperature of acquisition: room temperature; detector: scintillation counter detector and sample holder: non-rotating holder.
FT-IR spectroscopy (IR)
Fourier transform infrared analysis was performed on samples of crystalline and amorphous LUMF and (Soluplus, Kollidone VA64, Plasdone S630) melt extrudates of drug using a Fourier transform infrared spectrophotometer model 4100 (Spectrum GX-FT-IR, Perkin Elmer, USA). Samples were mixed with dry potassium bromide using a mortar and pestle, compressed to prepare a disk and analysed over a range 4,000–400 cm−1.
Scanning electron microscopy (SEM)
The shape and surface morphology of the LUMF powder and LUMF-loaded solid dispersion were examined using XL 30 Model JEOL 5400 scanning electron microscope made in japan during analysis. Double sided carbon tape was affixed on aluminium stubs over which powder sample of LUMF and prepared SD was sprinkled. The radiation of platinum plasma beam using JFC-1600 auto fine coater was targeted on aluminium stubs for its coating to make layer of 2 nm thickness above the sprinkled powder for 25 min. These prepared coated stubs were then placed in the vacuum chamber of a SEM and adjusted to maximum magnification to obtain excellent quality scanning images. Then, those samples were observed for morphological characterization using a gaseous secondary electron detector (working pressure: 0.8 Torr, acceleration voltage: 10–30.00 kV). SEM images were obtained at maximum and visible magnification to understand the surface interaction between drug and polymer.
In vitro dissolution studies
The LUMF solid dispersions were investigated for their dissolution behaviour, in the 1,000 ml of 0.1 N HCl buffer of pH 1.2 as dissolution medium at 37 ± 0.2 °C. A standardized basket technique was applied, using a USP dissolution apparatus I (Electrolab-DBK, Mumbai, India) at speed of 100 rpm. Quantity equivalent to 120 mg of LUMF was weighed and filled inside hard gelatine capsules and were used for the dissolution studies further. Aliquots of 10 mL were withdrawn from each vessel at predetermined time intervals (10, 20, 30, 40, 50 and 60 min), filtered over a cellulose acetate filter of 0.45 μ. At each time point, the same volume of fresh medium was replaced. The withdrawn 10 mL aliquot was further diluted to 10 times before UV-analysis. Samples were analysed using UV spectrophotometer for in vitro assessment of the amount of LUMF released from the SD by absorbance measurement at a wavelength of 338 nm. All experiments were performed in triplicate.
Moisture uptake and stability studies
Moisture uptake by drug and prepared solid dispersion was studied using Moisture balance MB 50C (Citizen, India). Moisture content of LUMF and SD was calculated in the form of %. After moisture content analysis LUMF and SD were placed in crucible at accelerated conditions of temperature 40 ± 2 °C and humidity 75 ± 5 % RH in environmental test chamber for 24 h (Thermo lab, India) (16). These samples were then analysed for drug content by UV spectroscopy. The method is useful to determine the effect of moisture on degradation of drug and prepared SD systems (Rasenack and Muller 2002).
Flowability of SD
The flowability of prepared SD was characterized by measuring angle of repose and Carr’s compressibility index (Kalogeras 2011). Angle of repose was determined by pouring the dispersion powder through a funnel (10 mm diameter orifice) onto a flat surface and measuring the angle between the horizontal and the slope of the heap of granules. Bulk density was calculated by measuring the volume of 10 g powder in a 10 mL cylinder. The cylinder was tapped 100 times until no further reduction in the volume of the SD powder was observed. Tapped density was calculated using the volume of the SD powder after tapping. Carr’s compressibility index (CI) was also determined.
Stability study
Stability studies were conducted by placing samples in closed glass vials which were stored in a controlled temperature environment inside stability chamber with relative humidity (RH) of 75 % and 40 °C temperature (Gavin and Osama 2010). Samples were removed after 6 months and tested for crystalline content using DSC and XRD. Drug release experiments were also conducted on samples stored for 6 months and compared with those tested immediately following manufacture. The assay of the SD was evaluated using high-performance liquid chromatography (HPLC). The mobile phase acetonitrile—0.1 M ammonium acetate buffer adjusted to pH 4.9 (85:15 %, v/v) was used as the mobile phases using HPLC as in case of pure LUMF. From SD, samples equivalent to 120 mg of LUMF were weighed dissolved in 5 mL of methanol and appropriately diluted and the drug content was determined by HPLC at λ = 338 nm.
Dissolution kinetic studies
Dissolution kinetic studies of prepared formulation were carried out using zero order, first order, Higuchi, Hixson Crowell and Korsemeyer Pappas equation model. Regression coefficient factor (r2) and other factors were calculated to understand the release kinetic performance of prepared SD formulations (Almeida and Possemiers 2011).
Antimalarial drug screening assay
The compounds were tested for in vitro antimalarial activity against Plasmodium falciparum 3D7 (chloroquine sensitive cell lines), ITG (chloroquine resistant cell lines) using the SYBR Green-I staining technique (Bacon et al. 2007 ).
SYBR green assay of plasmodium viability
Lysis buffer was made by adding Tris–HCl (20 mM; pH 7.5), EDTA (5 mM), Saponin (0.008 %w/v) and Triton X-100 (0.08 %v/v).
Standardization
Plasmodium falciparum culture was serially diluted with non-parasitized erythrocytes and medium to yield a haematocrit of 1 % and parasitemia levels ranging from 0 to 12 % to obtain a standard curve. Then a volume of 100 μL of the serially diluted culture was dispensed into a 96 well plate in triplicates, immediately followed by the addition of 100 μL of SYBR Green I in lysis buffer (0.2 μL of SYBR Green I/mL of lysis buffer). The plate was wrapped in aluminium foil and incubated on a shaker at RT for 30–60 min. The fluorescence was measured at 458 and 541 nm. The background fluorescence was subtracted for the empty well and the non-parasitized erythrocytes were analyzed by linear regression. A similar procedure was followed for the test samples. The 96 well micro plate was read using the HTS 7000 plus, bioassay reader (Perkin Elmer). The IC50 value was expressed as the drug concentration and various HME Formulations (F1 to F9) resulting in a 50 % inhibition of number of schizonts with three or more nuclei per 200 parasites by comparison with the drug-free control. The IC50 values for both methods were calculated by nonlinear regression analysis.
Molecular modelling interaction studies
The monomer unit structures of Soluplus, Kollidone VA64 or Plasdone S630 (Vinypyrrolidone-vinylacetate a common term was used during molecular modelling studies) and LUMF were constructed by using Gaussian programme in Schrodinger®, maestro software programme, USA. The energy minimization of individual as well as drug–polymer structure by calculating MM force field interaction was carried out. The docking was carried out to understand the bonding interactions between drug and polymer. The MD-simulation studies of stable molecules were run to understand the structural interaction mode and most stable confirmation of drug with each polymer (Maniruzzaman et al. 2013). In all the drug–polymer combination demonstrates strong hydrogen bonding interaction with up to distance of 4A° was observed. MD simulation gives the least energy and most stable confirmation of drug–polymer combination.
Results and discussion
Screening of polymers used in extrusion process
Amorphous drug substances are physically unstable due to their high energy state and tend to recrystallize upon storage. In order to stabilize these systems, various polymer carriers have been used because they readily generate amorphous forms and may be able to retain the amorphous nature of the drug upon storage. Various physical and chemical interactions of the drug and excipients in the extrudates were evaluated. In addition, long-term amorphous-state storage stability of the drug in HME dispersions was monitored at room and stability chamber temperature parameters to select polymers for extrusion process.
Soluplus (BASF product)
It is a polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer (Soluplus), a new polymer with amphiphilic properties was used. Soluplus shows exceptional solubilizing properties for BCS class II and class IV drugs, also offers the possibility of producing solid dispersions by hot-melt extrusion. The dissolution of poorly soluble drugs in aqueous media can be highly improved by the use of solid dispersions with Soluplus.
Kollidone VA64 (BASF product)
It is vinylpyrrolidone–vinyl acetate (60:40) copolymer specially used as binder in tablet formulations. It has excellent solubilizing properties in water and alcohol. Its application in the hot melt extrusion process has been investigated in current work.
Plasdone S630 (ISP product)
Plasdone S-630 copolymer is an excellent tablet binder, matrix polymer for solid dispersions and film former for topical applications. Plasdone S-630 copolymer is 60:40 linear random copolymers of N-vinyl-2-pyrrolidone and vinyl acetate.
Solubility parameters
The calculated solubility parameter for LUMF is 24.55 MPa1/2 and literature values for the polymers are 23.12, 22.55 and 22.94 MPa1/2 for Soluplus, Kollidone VA64 and Plasdone S-630, respectively (Osama and David 2012). Compounds with similar values for solubility parameters are likely to be miscible because the energy of mixing within the components is balanced by the energy released by the interaction between the components. It has also been postulated that compounds with a ∆δ (i.e. difference between δ values of two compounds) of less than 7.0 MPa1/2 are likely to be miscible, whereas compounds with a ∆δ of more than 10.0 MPa1/2 are likely to be immiscible. In this study, all polymers exhibited ∆δ of less than ±2 MPa1/2 compared to LUMD signifies drug-polymer miscibility in the SD formulations.
HPLC analyses
The method developed was found to be stable for acid, base, oxidation, reduction, heat and light degradation studies. Inter and intra coefficient of variation for LUMF was found to be in the range of ≤10 %.
Solubility studies
The solubility of the drug in the presence of concentrated solutions of a polymeric carrier can support to determine the mechanism of dissolution from a solid dispersion. To examine the solubilizing power of Soluplus, Kollidone VA64 and Plasdone S-630 the equilibrium solubility of crystalline LUMF in 0.1 N HCl of pH-1.2 comprising was determined and compared to the equilibrium solubility in the absence of polymer. Aqueous solubility of all the formulations were carried out and compared with that of pure LUMF. Solubility studies revealed the significant improvement in solubility of all SD formulation in water as well as in 0.1 N HCl. Solubility enhancement was found be in the order of LUM-Kollidon VA64 > LUM-Plasdone S630 > LUM-Soluplus (Fig. 2).

Saturation solubility studies of pure LUMF and SD formulation [mean ± SD (n = 3)]
Phase solubility studies
Phase inversion or transformation of phase of drug in polymeric environment with different concentration inside water was studied to understand the effect of SD when comes in contact with GI fluid. Also, absorption of drug with polymer inside body was predicted from this study. The results of phase solubility studies are shown in Fig. 3. The phase solubility is a function to examine the solubilising ability of polymer to solubilize drug in water in different concentration. As aqueous solubility is an important criterion for bioavailability of drug in GI fluid phase solubility studies has been carried out.

Phase solubility diagram of prepared SD in water [mean ± SD (n = 3)]
Theories and calculation
Gibbs free energy calculation
The ΔG°tr value provides information about whether the treatment is favourable or unfavourable for drug solubilization in an aqueous medium. Negative Gibbs-free energy values indicate improved dissolution and intermixing between drug and polymer. Negative values of Gibbs free energy indicates improved dissolution (Table 2).
Florey modelling results
It’s been reported that if coexistence χ ≥ 0.5/M, so there is presence of slightest degree of unfavourable interactions between the drug, polymer and excipient mixture which may cause phase separation. As shown in Table 3 the value of FH interaction factor is (χ) not more than or equal to 0.5/M. This is because the entropy of mixing is greatly reduced due to formation of molecular dispersion using HME. It indicates that the developed solid dispersions are thermodynamically stable and drug has been intermixed inside polymer matrix at molecular level. The obtained values of χ interaction parameter which are not ≥0.5/M, concluded the intermolecular miscibility between drug and polymer.
Gordon-Taylor calculations
Pure LUM in the DSC cycle showed a Tg of 131.54 °C and the amorphous polymers showed a Tg of 72.56, 92.95 and 94.79 °C for Soluplus, Kollidone VA64 and Plasdone S-630 based HME systems, respectively. A single Tg was observed for all the ratios of drug–polymer binary mixtures. This suggests the miscibility of drug and polymer in the given ratios and presence of a single phase in all the systems. According to the Gordon-Taylor equation, if the drug and polymer are miscible, the binary mixture will exhibit a single Tg(mix) that ranges between the Tg of the pure components and is dependent on the relative proportion of each component as shown in Table 4. The experimentally determined Tg of the binary mixtures is below the Tg of polymers, suggesting a antiplasticization effect of the drug on the polymer.
Characterization of extrudates
The optimized extrusion temperatures of 115 °C for Soluplus, 110 °C for Kollidon VA64 and 113 °C for Plasdone S-630 were used for processing the samples and it was observed that the polymer and drug were stable at these temperatures as determined by HPLC content analysis translucent extrudates were produced with all polymers at maximum 50 %wt./wt. drug loading. The drug content in the extrudates was at 99–102 % of the theoretical values as determined by HPLC assay (Gavin and Osama 2010).
DSC studies
The DSC thermograms show that the crystalline LUMF (Fig. 4) was characterized by a single, sharp melting endotherm at 131.54 °C (∆H = 14.64). DSC thermograms of fresh and aged SD are shown in Fig. 4. Disappearance of the melting endotherm in the DSC scan of SD suggested that the drug has been converted to the amorphous form during the extrusion process with DSC thermograms of used polymers. The pre-heated DSC thermograms of formulations F1, F4 and F7 (both fresh and 6 months) showed in Fig 5. Melt extrudate SD had no distinct melting endotherm for the LUMF which indicates the drug exists in the amorphous state in SD. The DSC analyses revealed complete conversion of crystalline LUMF into stable amorphous form. These results support the positive deviation of the experimental Tg values with the theoretically predicted values by the Gordon-Taylor equation.

DSC thermogram of pure LUMF, SD systems F1, F4, F7, F1 (aged), F4 (aged), F7 (aged) and polymers used

DSC thermogram of SD preheated cycle
XRD studies
The X-ray diffractograms are shown in Fig. 6 XRD of LUMF consist of sharp multiple peaks, indicating the crystalline nature of the drug with specific % crystallinity. In the case of SD (about 2 gm) when exposed to X-ray beam, shows disappearance of most of the crystalline characteristic peak intensities of LUMF. This indicates liable decrease in the crystalline nature of LUMF inside SD system which confirms amorphous nature of LUMF. In the XRD of LUMF peak intensities observed at (2θ) 4.51, 11.2, 14.65, 15.12, 18.12, 18.84, 20.31, 21.12, 22.89, 24.36, 25.78, 27.23, 28.91, 29.18 and 30.12. Characteristic peaks of LUMF observed at 5,000, 1,005, 3,085 and 18,824 related to its % crystallinity while these peaks disappeared in all SD systems. In the case of melt extrudates from F1, F4 and F7 intense peaks of LUMF vanished and percentage crystallinity also decreases substantially. While in the stability studies of melt extrudates from F1, F4 and F7 the intensity of LUMF characteristic peaks has disappeared or decreased to satisfactory amount after 6 months further confirms no recrystallization of LUMF in SD. From the XRD studies of both fresh and aged SD systems confirms the amorphous nature of LUMF with the polymers after HME.

PXRD diagrams of LUMF, SD (fresh and aged)
FTIR studies
Infrared spectroscopy has been mostly used to explore drug–polymer interactions in solid dispersion systems. In order to evaluate any possible chemical interactions between the drug and carriers, spectra of LUMF and HME formulations were examined Fig 7. IR spectrum of LUMF presented characteristic sharp peaks O–H stretching 3,419.03 cm−1, C–H stretching 2,934.42 cm−1, and C–O–O–C bending vibrations 1,084.51 cm−1, C=O stretching at 1,735.76 cm−1, OH bending at 1,333.94, CN stretch peak at 1,242.09, =C–H & –CH2 vibrations due to 875.55–973.64, C–H bending at 1,474.02 cm−1, C–Cl stretching (528.95 cm−1). IR spectrum of SD (F1) presented characteristic sharp peaks O–H stretching 3,750.44–3,466.04 cm−1, C–H stretching 2,953.29 cm−1, and C–O–O–C bending vibrations 1,072.01 cm−1, C=O stretching at 1,737.11 cm−1, OH bending at 1,373.88, CN stretch peak at 1,245.93, =C–H & –CH2 vibrations due to 879.95–932.92, C–H bending at 1,489.19 cm−1, C–Cl stretching (544.61 cm−1). IR spectrum of SD (F4) presented characteristic sharp peaks O–H stretching 3,377.23 cm−1, C–H stretching 2,951.57 cm−1, and C–O–O–C bending vibrations 1,088.70 cm−1, C=O stretching at 1,736.12 cm−1, OH bending at 1,374.46, CN stretch peak at 1,246.11, =C–H & –CH2 vibrations due to 879.42–933.69, C–H bending at 1,489.90 cm−1, C–Cl stretching (528.11 cm−1). IR spectrum of SD (F7) presented characteristic sharp peaks O–H stretching 3,398.07 cm−1, C–H stretching (2,937.87 cm−1), and C–O–O–C bending vibrations 1,152.5 cm−1, C=O stretching at 1,735.76 cm−1, OH bending at 1,333.94, CN stretch peak at 1,085.75, =C–H & –CH2 vibrations due to 875.12–973.71, C–H bending at 1,489.12 cm−1, C–Cl stretching (528.17 cm−1). However, IR spectra of SD (F1, F4 and F7) after 6 months show presence of all characteristic peaks signifies drug uniformity in terms of functional groups. The Soluplus exhibited CO stretch at 1,750 cm−1 and CH stretching (N-methylamine) at 2,750–2,850 cm−1, with Kollidone VA64 CO stretch at 1,710 cm−1, NH bending at 1,320 cm−1 and Plasdone S-630 spectra showed CO stretch at 1,680 cm−1. As shown in Fig. 7, the spectra of LUMF and HME formulations are identical. The LUMF skeleton stretching vibrations are not affected by the addition of polymer, suggesting no chemical interaction between the polymer and drug in the HME formulations. Soluplus has long chain hydroxyl groups favour the hydrogen bonding with LUMF and increase intermolecular interaction. Amphoteric nature of Soluplus makes the SD systems stable. The free –N and C=O groups forms hydrogen bonding with LUMF in HME formulations The Plasdone S-630 and Kollidone VA64 has two groups (=N and C=O) that can potentially form hydrogen bonds with LUMF in the HME formulations. The carbonyl group is more complimentary for hydrogen bonding and intermolecular interactions than the nitrogen atom because of steric hindrance. For HME formulations, the –OH stretching bands broadened and the intensity of the bands decreased, indicating some degree of interaction between the proton donating groups (–Cl and –OH) of LUMF and the proton accepting groups(C=O) in the polymers.

IR diagrams of fresh LUMF and SD systems F1, F4, F7, F1 (aged), F4 (aged), F7 (aged)
SEM analyses
SEM micrographs of pure LUMF and SD are shown in Fig. 8a–d. From the SEM micrograph it was evident that HME of LUMF resulted in a significant particle size reduction of LUMF. SEM micrographs of pure LUMF revealed large crystalline blocks, whereas SD was found to be without sharp edges. The SD appeared to be agglomerated with rough surface owing to the presence of polymer. Surface interaction between drug and polymer was observed at molecular level.

SEM microscopic images of pure a LUMF, b F1, c F4 and d F7
Dissolution studies
Because of the extreme low solubility of the drug, 1 % (w/v) Benzalkonium chloride (BKC) was added to the dissolution medium to maintain sink conditions. LUMF is a poorly soluble drug with a solubility 9.2 μg/mL in water. The saturation solubility of the LUMF was increased (be 97 μg/mL) by the addition of BKC to the dissolution medium. The increase in the dissolution rate in the case of the HME formulation is attributed to the amorphous state of the drug that which offers a lower thermodynamic barrier to dissolution and the formation of a glassy solution where the drug is molecularly dispersed in the polymer. The higher apparent solubility and increase in dissolution rate for amorphous materials is well known and has been extensively documented. The enhancement in solubility is the result of the disordered structure of the amorphous solid. Because of the short-range intermolecular interactions in an amorphous system, no lattice energy has to be overcome, whereas in the crystalline material, the lattice has to be disrupted for the material to dissolve. The solubility and dissolution rate of the drug were not enhanced by simple physical mixing with the polymer. Although BKC provided sufficient wetting of the drug particles as observed during dissolution studies, the polymers play crucial role in solubility enhancement.
The dissolution of the HME formulation with Soluplus (D30 = 89–96 %) was approximately 18-fold higher than LUMF alone. The dissolution of the Kollidone VA64-based HME formulation (D30 = 99–105 %) was approximately 21-fold higher than its corresponding LUMF alone (D30 = 5 %). The dissolution of the Plasdone S-630-based HME formulation (D30 = 93–100 %) was approximately 20-fold higher than its corresponding LUMF alone (D30 = 5 %). The enhancement in dissolution in Kollidone VA64 and Plasdone S-630 extrudates is also due to the conversion of crystalline drug into the amorphous state. The differences in the dissolution profile between the three polymer systems are due to the solubility/dissolution nature of the polymer in the dissolution medium. Dissolution of the drug in Soluplus is governed by the carrier, whereas in the case of Plasdone S-630 and Kollidone VA64 systems, the dissolution rate is governed by solubilization of the polymer to generate a hydrotropic condition for the insoluble drug. Thus, for Soluplus based systems, the dissolution is predominantly carrier controlled, whereas for Plasdone S-630 systems and Kollidone VA64, the drug dissolution is predominantly drug controlled. It was observed in the dissolution studies that Kollidone VA64 and Plasdone S630 of the SD formulation dissolved rapidly, leaving the drug as a fine precipitate. In the case of physical mixture, Kollidone VA64 and Plasdone S630 dissolved rapidly, leaving the crystalline drug in the dissolution medium. The high dissolution rate of LUMF from the Kollidone VA64 and Plasdone S630 is believed to be due to the drug–polymer microenvironment. Both have pH-independent solubility and dissolves rapidly; the pyrrolidone ring provides excellent solubility in water and a range of solvents, as well as adhesive, solubilization/crystal inhibition and film forming properties. The vinyl acetate in the polymer backbone lowers the polymer’s glass transition temperature (Tg) compared to polyvinylpyrrolidone (PVP) homopolymers and reduces hygroscopicity. As a result, copolymer is highly compressible, making it an excellent tablet binder. In addition, the unique combination of properties allows copolymer to improve solubility and bioavailability of poorly soluble drugs through the formation of melt extruded solid dispersions. Whereas Soluplus dissolves better in acid medium and rapidly because the pH at the polymer surface is increased when some Soluplus goes into solution, which retards the dissolution of the remaining undissolved polymer and subsequently makes the dissolution faster. Thus we found little advancement of dissolution rate in SD containing Kollidone VA64 and Plasdone S630 more as compared to Soluplus. Dissolution profiles of SD are shown in Figs 9 and 10.

Dissolution profile of SD formulations in 0.1 N HCl buffer of pH 1.2 [mean ± SD (n = 6)]

Dissolution profile of SD formulations in water [mean ± SD (n = 6)]
Moisture absorption studies
Hygroscopic nature of LUMF inside the SD formulation and pure LUMF was useful to understand the degradation effect of moisture. The SD were heated inside the chamber using UV-light source for 3 min at 100, 103 and 105 °C to calculate the moisture content in percentage which was displayed on screen. It was hypothesized that the extent of moisture absorption is directly proportional to the amount of hygroscopic surface area on the SD particles. Thus, moisture absorption would be indicative of the intimacy of mixing of LUMF with polymer matrix in SD. It also reveals the extent of complexation and coverage particle surface of drug in SD form. The Moisture content in SD found to be in the range of 1.02 ± 0.23 to 2.37 ± 0.24 % which is negligible and has no degradation effect on drug efficacy. The drug content analysis was carried out after treatment observed to be in the range of in the range of 97.86 ± 1.6 to 98 ± 1.2 % analysed by HPLC at 338 nm.
Powder flow characterization
Flow property calculation shows the excellent flow for prepared SD. It is important phenomenon for flow determination in making tablets or preparing capsules. Flow properties revealed easier scale up and feasibility of prepared SD using HME. The flow properties like bulk density and tapped density were carried out using standard procedures. Characteristic flow parameters like Angle of repose, Hausner’s ratio and Carr’s index were calculated and results indicated in Table 5.
Stability of SD
The dissolution stability was also evaluated for both initial and aged samples. Both HME formulations after storage were analogous to the preliminary formulations and did not show any melting endotherm as shown in the DSC thermograms. This indicated an amorphous state of the drug in the aged samples. The XRD results demonstrate similar diffractograms of aged as compared with fresh HME formulations, indicating the amorphous nature of the LUMF. Both DSC and XRD results on aged samples confirmed that there was no recrystallization of the amorphous drug in the SD formulations, suggesting good physical stability. The dissolution profiles of aged samples relative to fresh HME formulations further proved that the amorphous state of the drug was maintained in the aged formulations. The enhanced physical stability of the HME formulations upon storage is attributed to drug–polymer interactions and solubilizing effect of the polymer. The formulations were stable during 6 months period analysed by HPLC. Assay results of HPLC studies carried out represented in Table 6. Plasdone S-630 systems had strong intermolecular interactions, particularly hydrogen bonding between amorphous LUMF and the polymer. These might further reduce the molecular mobility and retarded recrystallization during storage.
Dissolution kinetic studies
The dissolution kinetic studies were carried out and the best suited results obtained in the case of Higuchi equation model as shown in Table 7. The value of r2 in Higuchi model is nearer to 0.1 and thus we conclude that dissolution followed Higuchi order kinetics.
In vitro antimalarial assay
In vitro antimalarial activity showed that SDs was active against P. falciparum 3D7 at a very low concentration. The IC50 value of SDs (0.084–0.213 ng/mL) was 220–101 times lower than the IC50 value of pure LUMF (18.2 ng/mL) and 45–18 times lower than the IC50 value of standard antimalarial drug, chloroquine (3.8 ng/mL). Antimalarial activity is found to be in the order of SD (Kollidon VA6) > SD (Plasdone S630) > SD (Soluplus) (Table 8)
Intermolecular interactions in melt extruded SD
After energy minimization of LUMF-Soluplus and LUMF-Vinypyrrolidone-vinylacetate strong hydrogen bonding interactions were identified. The stable confirmation with lowest energy values was optimised and MD simulation dynamics was started. The minimum interaction area from centroid was kept at 4A° to identify the possible interactions. The energy for stretching, bending, rotational, translational, torsional kinetic energies was calculated during this simulation process. The energy of combined system was found to be less than the addition of energies of individual molecules which signifies the improved stability of SD formulation. Hydrogen bonding interaction formed with the hydroxyl group of polymers with chlorine group of LUMF. Interactions are formed between the amine group of drug molecule and carbonyl groups of polymers. In both the polymers drug entrapment and interaction was favourable. Both hydroxyl and chlorine group within the LUMF molecule could form strong hydrogen bonds with the monomer of both polymers which signified by the optimal distance between the H-bond donor and acceptor. MD-simulation studies revealed the possible drug–polymer interactions. The energy calculations of LUMF and SD are shown in Table 9. The individual LUMF, polymer and stable MD confirmations are shown in Fig. 11a–e.

Energy minimised structures of a LUMF, b Soluplus and c Vinypyrrolidone-vinylacetate and MD simulation stable confirmations of d LUMF–Soluplus, e LUMF–Vinypyrrolidone–vinylacetate
Conclusion
Comparative evaluation using three solubilising polymers exposed the drug polymer miscibility characterised with the help of essential analytical techniques proves the productivity and industrial practicability of HME. Dissolution rate enhancement of LUMF was achieved by preparing amorphous glassy dispersions with Soluplus, Kollidone VA64 and Plasdone S-630 polymers by hot melt extrusion. The crystalline LUMF was converted to the amorphous state during the extrusion process with all polymers. Enhanced physical stability of the Soluplus, Kollidone VA64 HME and Plasdone S-630 formulation is attributed to drug–polymer interactions. Antimalarial activity of all SD formulation showed marked enhancement as compared to pure LUMF. MD simulation studies revealed possible molecular interaction between drug and polymer. Prepared bio-enhanced stable hot melt extruded LUMF powder SD systems using Soluplus, Kollidone VA64 and Plasdone S-630 would definitely enhanced therapeutic importance of LUMF as single drug candidate use, eventually solved the problem of poor solubility. In all Kollidone VA64 of BASF found to be superior to that of Plasdone S630 of ISP in terms of all solubility and dissolution behaviour, instead both are same chemically.
References
Almeida A, Possemiers S (2011) Ethylene vinyl acetate as matrix for oral sustained release dosage forms produced via hot-melt extrusion. Eur J Pharm Biopharm 77(2):297–305
Ambike AA, Mahadik KR, Paradkar A (2004) Stability study of amorphous valdecoxib. Int J Pharm 282:151–162
Andriantsoanirina V (2011) Resistance of Plasmodium falciparum to antimalarial drugs: impact on malaria pre-elimination in Madagascar. Med Trop Mars 1:298–304
Bacon DJ, Latour C, Picot S (2007) Comparison of a SYBR green i-based assay with a histidine-rich protein ii enzyme-linked immunosorbent assay for in vitro antimalarial drug efficacy testing and application to clinical isolates. Antimicrob Agents Chemother 51:1172–1178
Breitenbach J (2002) Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm 54:107–117
Chiou WL, Riegelman S (1971) Pharmaceutical applications of solid dispersion systems. J Pharm Sci 60:1281–1302
Chokshi RJ, Zia H, Sandhu HK, Shah NH, Malick WA (2007) Improving the dissolution rate of poorly water soluble drug by solid dispersion and solid solution. Drug Deliv 14(1):33–45
Crowley MM, Zhang F, Koleng JJ, McGinity JW (2002) Stability of polyethylene oxide in matrix tablets prepared by hot-melt extrusion. Biomaterials 23:4241–4248
Doshi DH, Betageri GV (1997) Carbamazepine and polyethylene glycol solid dispersion preparation, in vitro dissolution, and characterization. Drug Dev Ind Pharm 23:1167–1176
Ford JL (1986) The current status of solid dispersions. Pharm Acta Helvetica 61:69–88
Gahoia S, Jain GK (2012) Enhanced antimalarial activity of lumefantrine nanopowder prepared by wet-milling DYNO MILL technique. Colloids Surf B Biointerfaces. doi:10.1016/j.colsurfb.2012.01.047
Gavin PA, Osama AD (2010) Physicochemical characterization and drug-release properties of celecoxib hot-melt extruded glass solutions. J Pharm Pharmacol 62:1580–1590
Greenhalgh DJ, Timmins PY (1999) Solubility parameters as predictors of miscibility in solid dispersions. J Pharm Sci 88:1182–1190
Hancock BC, Zografi G (1997) Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci 86:1–12
Higuchi T, Connors K (1965) Phase-solubility techniques. Adv Anal Chem Inst Technol 4:117–130
Juma E (2008) A randomized, open-label, comparative efficacy trial of artemether-lumefantrine suspension versus artemether-lumefantrine tablets for treatment of uncomplicated Plasmodium falciparum malaria in children in western Kenya. Malar J 7:262–280
Kalogeras IM (2011) A novel approach for analyzing glass-transition temperature vs. composition patterns: application to pharmaceutical compound+polymer systems. Eur J Pharm Sci 42:470–483
Konno H, Taylor LS (2006) Influence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine. J Pharm Sci 95:2692–2705
Linn M (2012) Soluplus as an effective absorption enhancer of poorly soluble drugs in vitro. Eur J Pharm Sci 45:336–343
Makar RR, Latif R, Hosni EA, Omaima N, Gazayerly El (2013) Optimization for glimepiride dissolution enhancement utilizing different carriers and techniques. J Pharm Invest 43:115–131
Maniruzzaman M, Morgan DJ, Douroumis D (2013) Drug–polymer intermolecular interactions in hot-melt extruded solid dispersions. Int J Pharm 443:199–208
McGinity JW, Zhang F, Repka MA, Koleng JJ (2001) Hot-melt extrusion as a pharmaceutical process. Am Pharm Rev 4(2):25–36
Osama AA, David SJ (2012) Understanding the performance of melt-extruded poly(ethylene oxide)-bicalutamide solid dispersions: characterisation of microstructural properties using thermal, spectroscopic and drug release methods. J Pharm Sci 101:200–213
Rasenack N, Muller B (2002) Development of novel ibuprofen-loaded solid dispersion with improved bioavailability using aqueous solution. Arch Pharm Res 19:1894–1903
Repka MA, Prodduturi S, Stodghill SP (2003) Production and characterization of hot-melt extruded films containing clotrimazole. Drug Dev Ind Pharm 29:757–765
Suneela P (2005) Solid-state stability and characterization of hot-melt extruded poly(ethylene oxide) films. J Pharm Sci 94:2232–2245
Takeuchi H, Nagira S (2004) Solid dispersion particles of tolbutamide prepared with fine silica particles by the spray-drying method. Powder Technol 141:187–195
Vasconcelos T, Sarmento B, Costa P (2007) Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Disc Today 12:1068–1075
Zhang F, McGinity JW (1999) Properties of sustained- release tablets prepared by hot-melt extrusion. Pharm Dev Technol 4:241–250
Zsombor KN, Attila B (2012) Comparison of electrospun and extruded Soluplus -based solid dosage forms of improved dissolution. J Pharm Sci 101:322–332
Acknowledgments
The authors are thankful to Bajaj Healthcare Pvt. Ltd. (India) for providing the gift sample of Lumefantrine, BASF Ltd. Mumbai for the gift sample of Soluplus and Kollidone VA64 and, ISP Ltd. for Plasdone S630. The author is also thankful to UGC (SAP) for providing the research fellowship and Institute of Chemical Technology (Mumbai, India) for providing all facilities and guidance. The author is also thankful to Mr. Pavankumar Todkar from Dept. of Virology and Immunology, Haffkine Institute, Parel, Mumbai for helping with in vitro antimalarial studies at his laboratory.
Conflict of interest
Author declares that we do not have any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 1 (MP4 6826 kb)
Rights and permissions
About this article
Cite this article
Fule, R., Meer, T., Sav, A. et al. Solubility and dissolution rate enhancement of lumefantrine using hot melt extrusion technology with physicochemical characterisation. Journal of Pharmaceutical Investigation 43, 305–321 (2013). https://doi.org/10.1007/s40005-013-0078-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40005-013-0078-z