
Abstract
Microbes play an important role in carbon turnover in forest ecosystems by producing polysaccharide-degrading enzymes such as cellulase, xylanase, and β-glucosidase. In the present study, we isolated a bacterial strain producing cellulase and xylanase from the Forest Park at Gyeongnam National University of Science and Technology using LB agar plates containing 0.5 % carboxymethyl cellulose and 0.01 % trypan blue. Based on 16S rRNA sequencing and API analysis, the isolated strain was identified as a Bacillus species and named Bacillus sp. JYM1. The optimal growth temperature of Bacillus sp. JYM1 was 37 °C. The maximal activities of carboxymethyl cellulase (CMCase) and xylanase were obtained after a 24-h cultivation. The optimal pH and temperature were 6.0 and 50 °C for CMCase and 5.0 and 50 °C for xylanase, respectively. The gene responsible for the xylanase activity in Bacillus sp. JYM1 was cloned and expressed in Escherichia coli. The expressed recombinant protein showed similar biochemical properties to the xylanase of Bacillus sp. JYM1. Therefore, our results confirmed that the gene cloned from Bacillus sp. JYM1, herein named Bxyn, encodes xylanase.
Similar content being viewed by others


Introduction
The decomposition of organic matter as a carbon source is very important to maintain the balance of forest ecosystems because it supplies carbon for improving the soil properties and fertility (Waldrop et al. 2004; Ayres et al. 2009; Yang et al. 2014). Most of the carbohydrate in forest ecosystems is present as polysaccharides such as cellulose and hemicellulose, which form complexes with a recalcitrant matrix such as lignin in plant cell walls (Coughlan and Halzlewood 1993; Leonowicz et al. 2001). These polysaccharides can be degraded by microbes such as wood rot fungi or bacteria that secrete a large number of carbohydrate-active enzymes and polyphenol oxidase (Cantarel et al. 2014; Tolonen et al. 2011, 2015). Among them, species of the gram-positive genus Bacillus are the most well-known bacteria that secrete a variety of hydrolytic enzymes such as cellulases and hemicellulases to utilize various complex carbohydrates in their natural habitat (Schallmey et al. 2004; Kim et al. 2012). Therefore, Bacillus species have been considered to be very important for carbon cycling as well as the maintenance of healthy ecosystems because they degrade the organic matter in forest soils (Demain et al. 2005; Yang et al. 2014). In addition, Bacillus species are attractive organisms for industrial use because of their capacity for secreting proteins into the extracellular medium, and their fast growth rates that lead to short fermentation times (Duarte et al. 1999; Yasinok et al. 2010).
The enzymes related to the metabolism of plant carbohydrates can be classified into 133 families based on their amino acid sequence homology (http://www.cazy.org/Glycoside-Hydrolases.html) (Cantarel et al. 2014). Among them, the glycoside hydrolase (GH) families 5, 8, 9, 44, 48, and 61 are responsible for the cellulose-degrading activity (Cantarel et al. 2014). Cellulose, the most abundant biopolymer on earth, is a linear polymer chain with several hundred to many thousands of β-1,4-linked d-glucose units (Nishida et al. 2007; Back and Kwon 2007). Moreover, cellulose content generally ranges from 40 to 50 % in wood and is over 90 % in cotton fiber (Wang et al. 2006). The bioconversion of cellulose to smaller oligosaccharides or glucose is catalyzed by three major types of cellulases: endoglucanases (EC 3.2.1.4), exoglucanases (EC 3.2.1.91), and β-glucosidases (EC 3.2.1.21) (Cai et al. 1998, 1999). Endoglucanase, also called carboxymethyl cellulase (CMCase), randomly breaks down cellulose into shorter polysaccharides or oligosaccharides. Exoglucanase, also referred to as cellobiohydrolase, removes cellobiose or glucose from the non-reducing end of the cellulose chain. β-Glucosidase completely hydrolyzes cello-oligosaccharides and cellobiose into glucose (Annamalai et al. 2013).
Xylan, a group of plant hemicelluloses found in cell walls, is the second most abundant biopolymer next to cellulose. Xylan is a β-1,4-linked polymer of xylopyranosyl residues as the main chain with a degree of polymerization ranging from 70 to 200 (Whistler and Richards 1970). Xylans are typically 10–35 % of the hemicelluloses in hardwoods and 10–15 % of the hemicelluloses in softwood (Herbert 2006). Complete hydrolysis of the xylan polymers requires a wide variety of cooperatively acting GHs such as β-1,4-endoxylanases, β-xylosidases, α-glucuronidases, α-arabinofuranosidases, and esterases (Cantarel et al. 2014). Among the β-1,4-endoxylanases is the well-known GH xylanase, which is a crucial enzyme in the random hydrolysis of β-1,4-xylosidic linkages. Based on the amino acid sequence of their catalytic domains, microbial xylanases are currently categorized into six GH families (5, 8, 10, 11, 30, and 43) (Cantarel et al. 2014). These xylanases have been accepted as an environmentally friendly catalyst for hydrolyzing hemicellulosic polysaccharides to simple sugars, unlike chemical hydrolysis that can produce hazardous byproducts such as phenolic compounds, aliphatic acids, and furan derivatives (Palmqvist and Hahn-Hagerdal 2000). Therefore, xylanases have been used in a variety of food and industrial applications such as beer or juice production, animal feed processing, biofuel production, and pulp- and paper-related processes (Kulkarni et al. 1999; Ragauskas et al. 2006).
Bacillus species are attractive carbohydrate-degrading enzyme producers, owing to their ability to excrete large quantities of enzymes into the growth medium as well as their rapid growth rates compared with fungi and yeast (Tjalsma et al. 2004; Zhang and Zhang 2011). To understand the degradation of lignocellulosic materials such as plant roots and little falls by bacterial enzymes, a bacterial strain, Bacillus sp. JMY1, was isolated from forest soil. We determined the properties affecting the activities of CMCase and xylanase from Bacillus sp. JMY1. Furthermore, the Bxyn gene, which encodes xylanase, was cloned and biochemically characterized.
Materials and methods
Bacterial isolation
Soil samples were collected in March from Forest Park (FP, also called Jurassic Park), consisting of a mixed forest of broad-leaved (80 %) and coniferous (20 %) trees, located at the Gyeongnam National University of Science and Technology. Approximately 100 g of soil from the three regions was prepared from the top 10 cm using a small autoclaved spade, and the soil samples were passed through 2-mm sieves. Soil samples (1 g) were suspended in 10 mL of sterile distilled water and serially diluted up to 10−6 with sterile distilled water. The diluted samples were spread on LB agar plates containing 0.01 % trypan blue (Sigma-Aldrich, St. Louis, MO, USA) and 0.5 % carboxymethyl cellulose (CMC), and incubated at 28 °C for 2 days. The cellulolytic clones were selected based on the presence of clear halos around the colonies.
16S rRNA gene sequence and analysis
The 16S rRNA universal primers used for gene amplification and identification of the bacterial isolate were 877F: 5′-CGGAGAGTTTGATCCTGG-3′ as the forward primer and 878R: 5′-TACGGCTACCTTGTTAGCGAC-3′ as the reverse primer. The 16S rRNA gene was amplified by polymerase chain reaction (PCR) using the chromosomal DNA extracted with a Qiagen DNeasy kit (Hilden, Germany) according to the manufacturer’s instructions. The PCR reaction mixture was composed of 1 μL of template, 3 μL of 2.5 mM dNTPs, 3 μL of 10× reaction buffer, 3 μL of each primer (10 pmol/μL), 0.3 μL of nTaq-HOT polymerase (Enzynomics, Daejeon, Korea), and sterile distilled water to a final volume of 30 μL. A total of 30 cycles of PCR was performed under the following conditions: denaturation at 94 °C for 0.5 min, 50 °C for 1 min, and 72 °C for 1.5 min. PCR products were resolved by electrophoresis in 1.5 % agarose gels and purified with a DNA purification kit (Bioneer, Daejeon, Korea). The purified 16S rRNA gene was cloned into the pGEM-T Easy vector (Promega, Madison, WI, USA), and the sequence was analyzed. The sequence similarity and multiple sequence alignment of the 16S rRNA gene were analyzed by the BLSTN program provided by the National Center for Biotechnology Information (NCBI) and CLUSTAL W, respectively. Phylogenetic analysis of the 16S rRNA gene from the isolated Bacillus strain with other bacterial 16S rRNAs was performed using the neighbor-joining method with the Molecular Evolutionary Genetics Analysis 6.0 (MEGA 6.0) program (Tamura et al. 2013). Bootstrap analysis generated by 1000 sampling replicates was performed to assess the reliability of the branching patterns.
Zymogram assay
CMC-SDS-PAGE and xylan-SDS-PAGE were performed as described by Cho et al. (2006). After resolution of the enzyme samples by SDS-PAGE containing 0.4 % CMC and xylan, the protein was renatured by incubation with several changes of 200 mL of 10 mM sodium acetate buffer (pH 5.0) containing 1 % (v/v) Triton X-100 with a rotary shaker overnight at 45 °C. Subsequently, the gels were incubated in 10 mM sodium acetate buffer (pH 5.0) at 50 °C for 8 h (CMCase) or 14 h (xylanase). The gels were stained with 0.5 % (w/v) Congo red for 30 min at room temperature and destained with 1 M NaCl until pale-red hydrolytic zones appeared against a red background. Finally, 0.1 N HCl, which changes the background to dark blue, was used to facilitate photographic documentation.
Enzyme assay
The enzymatic activities of CMCase and xylanase using the culture supernatant of Bacillus sp. JYM1 were determined by measuring the amount of reducing sugars during incubation with CMC and xylan at 50 °C for 30 min. Briefly, 1 mL of reaction mixture containing 50 mM sodium acetate buffer (pH 5.0), 1 % (w/v) xylan or CMC, and 0.6 units of enzyme was incubated and boiled for 5 min after adding the same amount of 1 % dinitrosalicylic acid in order to determine the reducing sugar content. The released sugars were measured spectrophotometrically at 540 nm. One unit (U) of enzymatic activity was defined as the amount of enzyme releasing 1 µmol of reducing sugar per minute.
In order to determine the optimal temperature for CMCase and xylanase activity, the reaction mixture was incubated at different temperatures from 30 to 70 °C for 30 min. The thermal stabilities of CMCase and xylanase were compared by measuring the residual activities after pre-incubation of the enzyme at three different temperatures (40, 50, and 70 °C). The optimal pH for CMCase and xylanase was determined with 50 mM sodium acetate buffer (pH 3.0–6.0), 50 mM sodium phosphate buffer (pH 7.0–8.0), and 50 mM Tris–HCl buffer (pH 8.0–9.0).
Molecular cloning and expression of xylanase gene
The xylanase gene from Bacillus sp. JYM1 (Bxyn) was amplified from the genomic DNA by PCR. PCR was performed with nTaq-HOT DNA polymerase (Enzynomics) under the following conditions: a total of 35 cycles of 0.5 min at 95 °C for denaturation, 1 min at 55 °C for annealing, and 0.5 min at 72 °C for amplification. The primers, 5′-atGTCGACatgtttaagtttaaaaagaatt-3′ as the forward and 5′-atGCGGCCGCttaccacactgttacgttag-3′ as the reverse, were designed based on the published sequence of the xylanase gene from B. subtilis (Huang et al. 2006). In order to facilitate Bxyn subcloning for expression in E. coli, the restriction sites for SalI and NotI (which are capitalized in the primer sequences) were inserted in the forward and reverse primers, respectively. The PCR product was subcloned into the pGEM-T Easy vector (Promega) and the resulting Bxyn expression plasmid was sequenced.
To express Bxyn in E. coli, the Bxyn plasmid was digested with SalI and NotI and the Bxyn cDNA subcloned into the corresponding sites of the pGEX-5X-3 vector (GE Healthcare, Little Chalfont, UK). The resulting plasmid is referred to as PBxyn. The induction and purification of the recombinant Bxyn was performed as described by Min et al. (2002).
Results and discussion
Screening and identification of cellulolytic bacteria
To isolate cellulolytic bacteria from FP soil, we used the LB-CMC agar trypan blue method (Cho et al. 2006). Diluted soil samples were plated on the LB-CMC trypan blue agar plates. The number of total bacterial colonies was 1.96 × 107 colony-forming units (CFU)/g soil, and among those, 4.12 × 106 CFU/g soil were positive for cellulolytic activity, which accounts for about 21 % of the total culturable bacterial colonies. Although FP showed slightly lower numbers of culturable bacteria than Chang Qing Garden (CQG) in China, which consists of a 20-year-old forest planted with broad-leaved deciduous trees, the ratio of cellulolytic bacteria is higher compared to the 15 % determined for CQG (Yang et al. 2014). The difference in soil bacterial communities between FP and CQG is likely due to soil physicochemical parameters and the types of plant residues (Ulrich et al. 2008; Weinert et al. 2010; Wilson 2011; Yang et al. 2014).
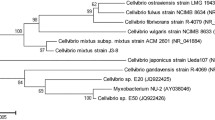
Among the cellulolytic bacteria, 20 strains showing relatively high activity were selected and subcultured on the LB-CMC trypan blue agar plates. From these 20 strains, one strain, which showed the highest activity on both CMC and xylan, was selected for further study. The isolated strain was first identified by 16S rRNA gene sequence analysis. The sequences were entered into the BLAST (NCBI) search engine and the identities were established. The highest identity for the isolated strain was with Bacillus spp. (99 %). Based on the API 50 analysis system, the isolated strain was determined to be Bacillus subtilis/amyloliquefaciens/atrophaeus with 97 % likelihood in the biochemical properties. Moreover, it belongs to the same clade as B. subtilis subsp. spizizenii NRRL, B. mojavensis IFO 15718T XH7, and Brevibacterium halotolerans LMG 21660T according to the phylogenetic analysis (Fig. 1). These results strongly support that the isolated strain is one of the Bacillus species. Therefore, the isolated strain was named Bacillus sp. JMY1.

A neighbor-joining phylogenetic tree based on the 16S rRNA gene sequence of Bacillus sp. JMY1. The Kimura two-parameter model was used to determine the distance matrix. The bootstrap values greater than 50 % based on 1000 replicates are listed as percentages at the branch points. Scale bar 0.05 substitutions per 100 nucleotides
Zymogram analysis of CMCase and xylanase
To identify the proteins responsible for CMCase and xylanase activities, the culture media and Bacillus sp. JMY1 cells were analyzed by SDS-PAGE containing 0.4 % CMC and birchwood xylan, respectively. After electrophoresis, the gels were renatured, stained with 0.5 % Congo red, and then destained with 0.5 % NaCl solution. Because the proteins with enzymatic activity degrade CMC and xylan, the active protein bands produced clear regions on the gel (Fig. 2). In the CMC zymogram, the cell pellet showed four active bands at about 55.3, 39.2, 38.7, and 27.2 kDa, whereas the culture filtrate (containing secreted proteins) showed corresponding bands at 39.2, 38.7, and 27.2 kDa (Fig. 2B). The B. subtilis HJ5 (CP007173.1) genome showed high similarity with Bacillus sp. JYM1 in the 16S rRNA analysis; it contained four endoglucanase homologue genes. In the signal peptide analysis using Signal P (http://www.cbs.dtu.dk/services/SignalP/), three of these homologues contain putative signal peptides for secretion into culture medium, but one of them with the predicted MW of 55.3 kDa does not have a signal peptide. This result indicates that the protein corresponding to 50 kDa would not be an extracellular cellulase. For xylanase, an active protein band produced a clear zone with an apparent molecular weight of 21–23 kDa in both the cell pellet and culture filtrate (Fig. 2C). Although the B. subtilis HJ5 (CP007173.1) genome contains four xylanase homologue genes, AKD35174.1 (1602 bp) AKD35236.1 (1269 bp) AKD35237.1 (1542 bp), and AKD35305.1 (642 bp), the AKD35305.1 (642 bp) gene might be the only one responsible for xylanase activity based on its predicted protein molecular weight of 23.4 kDa.

Detection of cellulase and xylanase activities from Bacillus sp. JMY1 by SDS-PAGE. Coomassie blue-stained SDS-PAGE gel (A), CMC zymogram (B), and xylan zymogram (C). M, molecular weight standard stained with Coomassie blue R-250; CP cell pellet, CF culture filtrate
Biochemical properties of cellulase and xylanase
To investigate the optimal growth temperature of Bacillus sp. JYM1, the strain was cultured at different temperatures (20, 25, 30, 37, and 45 °C). Although the optimal growth temperature of Bacillus sp. JYM1 was found to be 37 °C, it also grows well at different temperatures such as 25, 30, and 45 °C, but not at 20 °C (data not shown). To elucidate the relationship between enzymatic activity and culture duration, the activity of CMCase and xylanase in Bacillus sp. JYM1 was evaluated at different incubation times from 0 to 48 h. Both the bacterial growth and enzymatic activities of CMCase and xylanase gradually increased up to 24 h and then decreased (Fig. 3). The effect of temperature on CMCase and xylanase was measured from 20 to 70 °C. As a result, both CMCase and xylanase showed maximal activity at 50 °C, and the activity of both enzymes rapidly decreased at temperatures over 50 °C (Fig. 4A, C). CMCase showed more than 75 % of its activity at 30 and 40 °C (Fig. 4A). The optimal pH for CMCase and xylanase was determined in seven different buffers ranging from pH 3 to 9 at 50 °C using CMC and birchwood xylan as substrates, respectively. Although CMCase showed the highest activity at pH 6.0, it retained greater than 80 % of its activity at pH 4 and 5 (Fig. 4B). The optimal pH for xylanase was determined as pH 5.0, but it retained 92 % of its activity at pH 4.0 (Fig. 4D). To evaluate the thermal stability, the enzymes were incubated at 40, 50, and 70 °C without substrate for up to 60 min, and then their residual activities were measured. CMCase and xylanase were stable at 40 and 50 °C, respectively, and retained greater than 80 % of their original activities after a 60-min pre-incubation, while both activities rapidly decreased at 70 °C and retained less than 20 % of their original activities after a 30-min pre-incubation (Fig. 5). Finally, most activity losses for both enzymes were observed at 70 °C after a 30-min pre-incubation. These results suggest that CMCase and xylanase from Bacillus sp. JMY1 are mesophilic enzymes.

Cell growth and enzymatic activities of CMCase and xylanase according to culture time. Bacillus sp. JMY1 was cultured at 37 °C with shaking at 200 rpm and the enzymatic activities were determined in 50 mM sodium acetate buffer containing 1 % (w/v) CMC (pH 6.0) and xylan (pH 5.0) at 50 °C. All results are expressed as mean ± SD (n = 3)

Effects of temperature and pH on relative activity of CMCase and xylanase. Effect of temperature (A) and pH (B) on CMCase activity. Effect of temperature (C) and pH (D) on xylanase activity. The effects of temperature and pH were determined in 50 mM sodium acetate buffer containing 1 % (w/v) CMC and xylan, respectively at 50 °C. All results are expressed as mean ± SD (n = 3)

Thermal stability of CMCase (A) and xylanase (B) from Bacillus sp. JMY1. The thermal stability was determined in 50 mM sodium acetate buffer containing 1 % (w/v) CMC (pH 6.0) and xylan (pH 5.0) at 40, 50, and 70 °C. All results are expressed as mean ± SD (n = 3)
Isolation and characterization of the Bacillus sp. JYM1 xylanase gene
Two primers, which were designed based on the published sequence of the xylanase gene from B. subtilis (Huang et al. 2006), were used to obtain the full-length gene encoding xylanase. The PCR product was then subcloned and sequenced, and the resulting gene was named Bxyn. The Bxyn gene, which contains a 642-bp open reading frame, was predicted to encode a protein with a molecular mass of 23.4 kDa and a theoretical pI of 9.32 (Fig. 6). A putative signal peptide cleavage site determined by Signal P analysis (http://www.cbs.dtu.dk/services/SignalP/) and the SMART program (Schultz et al. 2000) was found between amino acids 28 and 29. Blast analysis using the NCBI BLAST tool showed that the deduced amino acid sequence was highly similar to that of endo-β-1,4-xylanase and contained a catalytic domain characteristic of GH11 enzymes and no carbohydrate-binding domain from other bacteria, especially Bacillus species. In Bxyn, the putative catalytic residues Glu-106 and Glu-200, which perform the double-displacement reaction of retaining GH enzymes (MacLeod et al. 1994), were conserved (Fig. 6).

Nucleotide and deduced amino acid sequences of xylanase from Bacillus sp. JMY1. The nucleic acid sequence of the xylanase gene (Bxyn) is presented on the top line, and the deduced amino acid sequence is shown below. The stop codon is marked with an asterisk. The signal peptide and GH11 domain predicted by the SMART program (Schultz et al. 2000) are indicated by a black bar and solid-line box, respectively. The catalytic residues, Glu-106 and Glu-200, are marked by down arrow
The Bxyn gene was cloned and expressed in E. coli BL21 (DE3) to characterize the protein’s biochemical properties. Bxyn was expressed as an N-terminal glutathione S-transferase (GST) fusion protein to facilitate its expression and purification. The Bxyn recombinant proteins were purified by GST-affinity column chromatography and analyzed by SDS-PAGE. Two bands, 49.4 and 26 kDa, were observed (Fig. 7A). The protein with a molecular weight of 49.4 kDa was the recombinant Bxyn (23.4 kDa) fused with the GST (26 kDa). The 26 kDa protein was likely GST that had been cleaved from the recombinant Bxyn by endogenous E. coli protease(s). The soluble Bxyn recombinant protein exhibited xylanase activity of 0.41 U/mg protein, whereas E. coli extracts harboring the empty pGEX-5×-3 vector did not display xylanase activity, confirming that the Bxyn gene encodes xylanase. Although the optimal pH of Bxyn was determined to be pH 5.0, it showed 75 % of its maximal activity at the pH range of 4.0–8.0 (Fig. 7B). Although the optimal temperature of Bxyn at pH 5.0 was 50 °C, about 70 % of the maximal activity was detected at 60 °C. Consistent with the results of the previous section, the activity of the recombinant Bxyn was reduced to about 30 % at 70 °C (Fig. 7C). The thermal stability of recombinant Bxyn was also evaluated and the result was similar to that for native Bxyn in Bacillus sp. JYM1 (Fig. 7D). Additionally, the CMCase activity of Bxyn was evaluated, but there was no detectable CMCase activity as expected. The effects of different metal ions on the purified Bxyn were evaluated. Bxyn activity was slightly enhanced by Fe2, Mg2+, Cu2+, and the metal chelator ethylenediaminetetraacetic acid (EDTA), but not Ca2+ and Zn2+ (Table 1). In previous studies, Cu2+ strongly inhibited the activity of several xylanases (Araki et al. 1999; Fialho and Carmona 2004; Gupta et al. 2000; Liu et al. 1999) and is therefore considered a major problem for industrial application of xylanase. However, as reported by Guo et al. (2009) and Khandeparket et al. (2011), Bxyn was virtually unaffected by copper. Although the activity of several xylanases such as those from Aspergillus niger (Krisana et al. 2005), Aspergillus giganteus (Fialho and Carmona 2004), and B. subtilis, cho 40 (Khandeparker et al. 2011) was reported to be inhibited by EDTA, Bxyn, similarly to Bacillus pumilus ARA xylanase (Qu and Shao 2011), which was not inhibited by EDTA, indicating that it is not a metalloenzyme.

SDS-PAGE analysis, effects of pH, temperature, and thermal stability of Bxyn. (A) Coomassie blue-stained SDS-PAGE. M molecular weight standards, BI before induction, AI after IPTG induction, P the purified Bxyn. (B) Effect of pH on Bxyn activity. (C) Effect of temperature on Bxyn activity. (D) The thermal stability of Bxyn activity
In summary, recombinant Bxyn is applicable to the pulp industry as well as the animal feed industry because of its relatively broad pH and temperature range, as well as its ability to maintain maximal enzymatic activity in the presence of copper (Viikari et al. 1994; Beg et al. 2001; Collins et al. 2005).
References
Annamalai N, Rajeswari MV, Elayaraja S, Balasubramanian (2013) Thermostable, haloalkaline cellulase from Bacillus halodurans CAS 1 by conversion of lignocellulosic wastes. Carbohydr Polym 94(409):415
Araki T, Tani S, Maeda K, Hashikawa S, Nakagawa H, Morishita T (1999) Purification and characterization of β-1, 3-xylanase from a marine bacterium, Vibrio sp. XY-214. Biosci Biotechnol Biochem 63:2017–2019
Ayres E, Steltzer H, Berg S, Wall DH (2009) Soil biota accelerate decomposition in high-elevation forests by specializing in the breakdown of litter produced by the plant species above them. J Ecol 97:901–912
Back SC, Kwon YJ (2007) Optimization of the pretreatment of rice straw hemicellulosic hydrolyzates for microbial production of xylitol. Biotechnol Bioprocess Eng 12:404–409
Beg QK, Kapoor M, Mahajan L, Hoondal GS (2001) Microbial xylanase and their industrial application: a review. Appl Microbiol Biotechnol 56:326–338
Cai YJ, Buswell JA, Chang ST (1998) β-Glucosidase components of the cellulolytic system of the edible straw mushroom, Volvariella volvacea. Enzym Microb Technol. 22:122–129
Cai YJ, Chapman SJ, Buswell JA, Chang ST (1999) Production and distribution of endoglucanase, cellobiohydrolase, and beta-glucosidase components of the cellulolytic system of Volvariella volvacea, the edible straw mushroom. Appl Environ Microbiol 65:553–559
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495
Cho KM, Hong SY, Lee SM, Kim YH, Kahng GG, Kim H, Yun HD (2006) A cel44C-man26A gene of endophytic Paenibacillus polymyxa GS01 has multi-glycosyl hydrolases in two catalytic domains. Appl Microbiol Biotechnol 73:618–630
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29:3–23
Coughlan MP, Hazlewood GP (1993) Beta-1,4-D-xylan-degrading enzyme systems: biochemistry, molecular biology and applications. Biotechnol Appl Biochem 3:259–289
Demain AL, Newcom M, Wu JHD (2005) Cellulase, clostridia, and ethanol. Microbiol Mol Bio Rev 69:124–154
Duarte MCT, Portugal EP, Ponezi AN, Bim MA, Tagilari CV, Franco TT (1999) Production and purification of alkaline xylanases. Bioresour Technol 68:49–53
Fialho MB, Carmona EC (2004) Purification and characterization of xylanase from Aspergillus giganteus. Folia Microbiol 49:13–18
Guo B, Chen XL, Sun CY, Zhou BC, Zhang YZ (2009) Gene cloning, expression and characterization of a new cold-active and salt-tolerant endo-beta-1,4-xylanase from marine Glaciecola mesophila KMM 241. Appl Microbiol Biotechnol 84:1107–1115
Gupta S, Bhushan B, Hoondal GS (2000) Isolation, purification and characterization of xylanase from Staphylococcus sp. SG-13 and its application in biobleaching of kraft pulp. J Appl Microbiol 88:325–334
Herbert S (2006) Handbook of pulp 1. Wiley, Weinheim, pp 28–30
Huang J, Wang G, Xiao L (2006) Cloning, sequencing and expression of the xylanase gene from a Bacillus subtilis strain B10 in Escherichia coli. Bioresour Technol 97:802–808
Khandeparker R, Verma P, Deobagkar D (2011) A novel halotolerant xylanase from marine isolate Bacillus subtilis cho40: gene cloning and sequencing. N Biotechnol 28:814–821
Kim YK, Lee SC, Cho YY, Oh HJ (2012) Isolation of cellulolytic Bacillus subtilis strains from agriculrural environments. ISRN Microbiol Article ID 650563: 9
Krisana A, Rutchadaporn S, Jarupan G, Lily E, Sutipa T, Kanyawim K (2005) Endo-1, 4-β-xylanase B from Aspergillus cf. niger BCC14405 isolated in Thailand: purification, characterization and gene isolation. J Biochem Mol Biol 38:17–23
Kulkarni N, Shendye A, Rao M (1999) Molecular and biotechnological aspects of xylanases. FEMS Microbiol Rev 23:411–456
Leonowicz A, Cho NS, Luterek J, Wilkolazka A, Wojtas-Wasilewska M, Matuszewska A, Hofrichter M, Wesenberg D, Rogalski J (2001) Fungal laccase: properties and activity on lignin. J Basic Microbiol 41:185–227
Liu R, Qu Y, Jiang Y, Gao P (1999) Purification and characterization of alkaline xylanases from Pseudomonas G6–2. Wei Sheng Wu Xue Bao 39:132–136
MacLeod AM, Lindhorst T, Withers SG, Warren RAJ (1994) The acid/base catalyst in the exoglucanase/xylanase from Cellulomonas fimi is glutamic acid 127: evidence from detailed kinetic studies of mutants. Biochemistry 33:6371–6376
Min SY, Kim BG, Lee C, Hur HG, Ahn JH (2002) Purification, characterization, and cDNA cloning of xylanase from Fungus Trichoderma strain SY. J Microbiol Biotechnol 12:890–894
Nishida Y, Suzuki KI, Kumagai Y, Tanaka H, Inoue A, Ojima T (2007) Isolation and primary structure of a cellulase from the Japanese sea urchin Strongylocentrotus nudus. Biochimie 89:1001–1002
Palmqvist E, Hahn-Hagerdal B (2000) Fermentation of lignocellulosic hydrolyzates. II. Inhibitors and mechanism of inhibition. Bioresour Technol 74:25–33
Qu W, Shao W (2011) Cloning, expression and characterization of glycoside hydrolase family 11 endoxylanase from Bacillus pumilus ARA. Biotechnol Lett 33:1407–1416
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ Jr, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T (2006) The path forward for biofuels and biomaterials. Science 311:484–489
Schallmey M, Singh A, Ward OP (2004) Developments in the use of Bacullus species for industrial production. Can J Microbiol 50:1–17
Schultz J, Copley RR, Doerks T, Ponting CP, Bork P (2000) SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res 28:231–234
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax WJ, Kuipers OP, Bron S, Hecker M, van Dijl JM (2004) Proteomics of protein secretion by Bacillus subtilis: separating the "secrets" of the secreome. Microbiol Mol Biol Rev 68:207–233
Tolonen AC, Haas W, Chilaka AC, Aach J, Gygi SP, Church GM (2011) Proteome-wide systems analysis of a cellulosic biofuel-producing microbe. Mol Syst Biol 7:461. doi:10.1038/msb.2010.116
Tolonen AC, Cerisy T, El-Sayyed H, Boutard M, Salanoubat M, Church GM (2015) Fungal lysis by a soil bacterium fermenting cellulose. Environ Microbiol 17:2618–2627
Ulrich A, Klimke G, Wirth S (2008) Diversity and activity of cellulose decomposing bacteria, isolated from a sandy and a loamy soil after long-term manure application. Microb Ecol 55:512–522
Viikari L, Kantelinen A, Sundquist J, Linko M (1994) Xylanases in bleaching: from an idea to the industry. FEMS Microbiol Rev 13:335–350
Waldrop MP, Zak DR, Sinsabaugh RL, Gallo M, Lauber C (2004) Nitrogen deposition modifies soil carbon storage through changes in microbial enzymatic activity. Eco Appl 14:1172–1177
Wang S, Cheng Q, Rials TG and Lee S-H (2006) Cellulose microfibril/nanofibril and its nanocompsites. In: Proceedings of the 8th Pacific rim bio-based composites symposium
Weinert N, Meincke R, Gottward C, Heuer H, Schloter M, Berg G, Smalla K (2010) Bacterial diversity on the surface of potato tubers in soil and the influence of the plant genotype. FEMS Microbiol Ecol 74:114–123
Whistler RL, Richards EL (1970) Hemicelluloses. In: Pigman W, Horton D (eds) The carbohydrates, vol 173. Academic press, New York, pp 697–703
Wilson DB (2011) Microbial diversity of cellulose hydrolysis. Curr Opin Microbiol 14:1–5
Yang JK, Zhang JJ, Yu HY, Cheng JW, Miao LH (2014) Community composition and cellulase activity of cellulolytic bacteria from forest soils planted broad-leaved deciduous and evergreen trees. Appl Microbiol Biotechnol 98:1449–1458
Yasinok AE, Brian S, Kocabas A, Bakir (2010) Xylanase from a soil isolate, Bacillus pumilus: gene isolation, enzyme production, purification, characterization and one-step separation by aqueous-two-phase system. World J Microbiol Biotechnol 26:1641–1652
Zhang XZ, Zhang YHP (2011) Simple, fast and high-efficiency transformation system for directed evolution of cellulase in Bacillus subtilis. Microb Biotechnol 4:98–105
Acknowledgments
This study was supported by Gyeongnam National University of Science and Technology in 2014.
Author information
Authors and Affiliations
Corresponding author
Additional information
Chong Kyu Lee and Min-Yeong Jang have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Lee, CK., Jang, MY., Park, H.R. et al. Cloning and characterization of xylanase in cellulolytic Bacillus sp. strain JMY1 isolated from forest soil. Appl Biol Chem 59, 415–423 (2016). https://doi.org/10.1007/s13765-016-0179-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13765-016-0179-2