
Abstract
Finding a green approach for silver nanoparticle (AgNP) synthesis and as an adsorbent for safe contaminant removal in water has become a challenge. In this study, AgNP were green synthesised into a biopolymer matrix (natural rubber latex/polyvinyl alcohol (PVA/NRL)) via electrospinning. Efficiency of the synthesised nanofibre composites in removing mercury (Hg) from an aqueous solution was examined. SEM and TEM analysis showed that AgNP were more visible on the fibre’s surface at a lower concentration (0.01 M AgNP), whilst at a higher concentration (0.015 M AgNP), relatively more AgNP were found within the fibre. The average sizes of the AgNP were relatively larger (8.15 ± 0.09 nm) in 0.01 M AgNP than in 0.015 M AgNP (2.82 ± 0.04 nm). FT-IR and XRD analyses confirmed the formation of AgNP in the PVA/NRL nanofibre composites. Maximum Hg adsorbed by the nanofibre composites occurred at pH 7 and 60 min. Experimental data were tested with different adsorption isotherms (Langmuir, Freundlich and Dubinin-Radushkevich) and kinetics models (pseudo-first order, pseudo-second order, Elovich and intraparticle diffusion equations). The results indicated that 0.01 M AgNP nanofibre composite fitted best to Langmuir isotherm, whilst 0.015 M AgNP fitted best to Dubinin-Radushkevich isotherm. Both nanofibre composites fitted best to the Elovich kinetic model. In general, 0.01 M AgNP (qmax = 68.3223 mg/g) was more efficient in removing mercury from the aqueous solution than 0.015 M AgNP (qmax = 26.6855 mg/g) within the same time frame. Therefore, a high potential exists for AgNP-PVA/NRL composite produced via green synthesis and electrospinning to remove mercury from aqueous solution, as shown in this study.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mercury (Hg) contamination of water has gained worldwide attention due to its non-biodegradable, bioaccumulative and highly toxic nature to living organisms. Mercury is the only liquid metal at room temperature and exists in elemental, inorganic and organic forms in the environment (Balali-Mood et al. 2021; Bernhoft 2012). Although Hg occurs naturally, anthropogenic activities such as Hg usage in pesticides, manufacturing fluorescent bulbs, batteries and illegal artisanal and small-scale gold mining have contributed to high levels in the environment (Balali-Mood et al. 2021; Diaz Arriaga 2015; Esdaile and Chalker 2018; Tchounwou et al. 2012). Primary sources of human exposure to Hg are inhalation of Hg vapour and consuming fish contaminated with Hg (Park and Zheng 2012; Tchounwou et al. 2012). Even though Hg vapour from anthropogenic sources can be controlled by limiting its exposure to elevated temperatures, methyl mercury (CH3Hg+) can only be controlled by preventing Hg release into water bodies or removing Hg from the water before methylation. Elemental Hg is converted to CH3Hg+ by algae and bacteria through methylation, which ends up in humans after consuming fish contaminated with Hg (Tchounwou et al. 2012). Even at low concentrations, exposure to Hg can lead to chronic effects, resulting in bioaccumulation in humans (Park and Zheng 2012). Mercury obtained via eating fish is easily absorbed by the gastrointestinal tract. Adsorbed Hg can cross the blood–brain barrier and placenta and can also accumulate in the kidney, neurological tissues and liver. All forms of Hg are toxic and can have detrimental neurological, gastrointestinal and nephrological effects (Balali-Mood et al. 2021; Tchounwou et al. 2012).
Removing Hg at low concentrations from wastewater is problematic, especially in countries where Hg releases are poorly regulated. Methods such as filtration, chemical precipitation as hydroxides, ion exchange and adsorption are used to remove Hg from wastewater; however, most of these methods are expensive, time-consuming and inefficient (Ganzagh et al. 2016). In recent studies, adsorption using silver nanoparticles (AgNP) has proven efficient in removing Hg from water, even at low concentrations (El-Tawil et al. 2019; Ganzagh et al. 2016; Katok et al. 2012; Korobeinyk and Inglezakis 2018). In solution, AgNP do not oxidise; however, AgNP forms a stable Ag-Hg amalgam (Katok et al. 2012). Therefore, AgNP as an adsorbent will be efficient in removing Hg from wastewater before methylation by algae and bacteria for fish to bioaccumulate.
Another pressing problem is the synthesis of AgNP using synthetic capping and reducing agent such as sodium borohydride, which is harmful to the environment (Badi’ah et al. 2019; Li et al. 2012). This plight has led researchers to seek green methods such as using corn starch (Kumar et al. 2018; Salaheldin 2018), leaf extracts (Ajitha et al. 2015; Al-Qahtani 2017; Jha and Prasad 2010; Mohammadi et al. 2019) and fruit extracts (Moldovan et al. 2016) to synthesise AgNP.
Natural rubber, a renewable resource widely cultivated in West Africa (including Ghana), Central Africa and Southeast Asia (Mooibroek and Cornish 2000; Venkatachalam et al. 2013), has proven efficient in synthesising AgNP (Abu Bakar et al. 2007, 2010; Danna et al. 2016; Guidelli et al. 2011; Suwatthanarak et al. 2016). Danna et al. (2016), Guidelli et al. (2011) and Abu Bakar et al. (2007) pointed out that proteins in natural rubber serve as a reducing and capping agent in AgNP synthesis. In its latex form, natural rubber can be mixed with polyvinyl alcohol (PVA), and without using synthetic solvents, it can be electrospun into nanofibres that can house AgNP for water treatment (Osei et al. 2022). In this form, issues on nanoparticles forming part of the contamination problem after water treatment can be addressed efficiently.
The possibility of synthesising green AgNP in a biopolymer matrix such as natural rubber latex/polyvinyl alcohol (PVA/NRL) via an electrospinning process has not been explored. Also, the potential of synthesised AgNP in PVA/NRL nanofibre composite to adsorb metals like Hg has not been investigated. Therefore, the present work seeks to investigate the possibility of synthesising AgNP into a PVA/NRL matrix via electrospinning and to evaluate the capacity of this nanofibre composite for Hg removal from an aqueous solution. The main parameters considered in the sorption experiments were pH, contact time and initial Hg concentration.
Materials and methods
Materials
Ammonium hydroxide (NH4OH, 26.5%; Deajung Chemical and Metals Co. Ltd.) was used as an ammonia (NH3) source. Natural rubber latex (NRL) from Hevea brasiliensis was collected from a rubber plantation at Bepo in the Western Region of Ghana, and stabilised using 0.7% (w/w) NH3. Powdered PVA (MW = 105.6 kDa, alcoholysis degree = 88%; Eastchem) was used to increase NRL’s spinnability. Silver nitrate (AgNO3; Merck) was used as a silver (Ag) source for AgNP synthesis, and solutions for the sorption experiment were prepared from Hg stock solution (1000 mg/L; Sigma-Aldrich). Nitric acid (HNO3, 68%; Brenntag Ghana Ltd.) and sodium hydroxide (NaOH, 97%; Paskem Fine Chem Pvt. Ltd.) were also used. Hydrochloric acid (HCl, 25%; Merck) was used to adjust the pH of solutions for point of zero charge (PZC) analysis, and sodium chloride (NaCl, 99.5%; Paskem Fine Chem Pvt. Ltd.) was used as a salt for PZC analysis.
Synthesis and electrospinning AgNP-NRL/PVA nanofibre composite
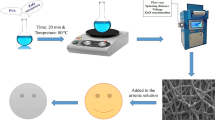
A 15% (w/v) PVA solution was mixed with NRL to form a 50% (w/v) PVA/NRL solution. The 50% ratio of PVA to NRL was selected as the composition for the wall material for AgNP synthesis. This was based on an earlier study by Osei et al. (2022).
The AgNP were fabricated into the PVA/NRL nanofibres by adding AgNO3 to the solution before electrospinning. Solutions of 0.10 M and 0.015 M AgNO3 were added to the PVA/NRL solution to form 0.01 M and 0.015 M AgNO3-PVA/NRL solutions, respectively. Prepared solutions were electrospun to form nanofibres, namely 0.01 M AgNP and 0.015 AgNP. This was done using an electrospinning unit (TL-01, Tongli Tech) with an applied voltage, flow rate, spinneret size, sliding unit speed, travel distance, collector (diameter = 76 mm) speed, relative humidity and temperature of 24 kV, 1.0 mL/h, 20 G, 5 mm/s, 23 cm, 1000 rpm, 40% and 30 ℃, respectively. The spinneret tip was kept 15 cm from the collector, and 8 mL of the prepared solution was dispensed for each nanofibre. The fibres were collected on an aluminium foil and were dried overnight at 50 ℃ in an oven.
Characterisation of AgNP-PVA/NRL nanofibre composite
Prepared nanofibre composites were first imaged using scanning electron microscopy (SEM; Zeiss EVO MA 15) to investigate the morphology and how the AgNP were dispersed on the surface of the nanofibre composite at 20 kV. The nanofibre composites were sputter-coated with a gold/palladium alloy to serve as a conductive layer before imaging. Next, the elemental composition of AgNP nanofibre composites was analysed using energy dispersive x-ray spectroscopy (EDS; Bruker). Transmission electron microscopy (TEM; FEI Tecnai T20) was also employed to image the AgNP on and within the fibre matrix, and ImageJ software was used to determine the average size of AgNP (based on 1206 particles for each nanofibre composite). Fourier transform-infrared (FT-IR; Bruker ALPHA Platinum-ATR) spectroscopy was employed in determining the surface functionalisation of the nanofibre composite. Due to the thin and elastomeric nature of the nanofibre composite, the fibre films were kept on the aluminium foil, and their crystalline properties were analysed by x-ray diffractometry (XRD; Malvern Panalytical Empyrean).
The point of zero charge (PZC) of both nanofibre composites were also determined to understand the surface charge of the composites during adsorption studies. The PZC was conducted with minor modifications, as Nasiruddin Khan and Sarwar (2012) reported. A 0.50 M HCl and 0.05 M NaOH were used to adjust the pH of 0.05 M NaCl to between pH 3 and 10. The nanofibre composites were placed into the NaCl solution and agitated, after which the final pH was determined.
Sorption experiment
Adsorption experiments were performed by placing 5 × 5 cm2 nanofibre composite in 50 mL of Hg solution. The solution was agitated on an orbital shaker at 200 rpm and at a temperature of 27 ℃. After agitation at a predetermined interval, samples of the solution were analysed for Hg concentration using atomic absorption spectrophotometry (AAS; Shimadzu AA 7000) hydride technique. The effect of contact time (0–120 min), pH (4–9) and initial concentration (0.01–1 mg/L) of Hg solution on the amount of Hg adsorbed were studied. The amount of Hg adsorbed by nanofibre composite and its removal efficiency (RE) were estimated using Eqs. 1 and 2, respectively.
where qe (mg/g) is the amount of adsorbate adsorbed at equilibrium; Co (mg/L) and Ce (mg/L) are the solution’s initial and equilibrium adsorbate concentration, respectively; V (L) is the volume of the adsorbate solution; and m (g) is the mass of the adsorbent. For a travel distance of 23 cm, a 549.1504 cm2 area film was produced with a Ag mass of 8.6294 mg and 12.9442 mg for 0.01 M and 0.015 M AgNO3 solutions, respectively. Therefore, every 5 × 5 cm2 nanofibre composite of 0.01 M and 0.015 M AgNO3 contained ~ 0.3929 mg and ~ 0.5893 mg of Ag, respectively.
Adsorption isotherms
Adsorption isotherm models were employed to examine the relationship between the concentration of adsorbates in the solution and the adsorbent (Sawyer et al. 2003; Tran et al. 2016). In this study, Langmuir, Freundlich and Dubinin-Radushkevich (D-R) isotherm models commonly used in analysing adsorption data were investigated (Bagal and Raut-Jadhav 2021). Langmuir isotherm assumes that adsorbates bind to the adsorbents in a monolayer fashion and that all active sites of the adsorbent have the same affinity for the adsorbate (Sawyer et al. 2003; Tran et al. 2017). The Langmuir model used is presented in Eq. 3 (Langmuir 1918).
where qmax (mg/g) is the maximum adsorption capacity of the adsorbent and KL (L/mg) is the measure of affinity between the adsorbate and adsorbent or Langmuir constant. In the Langmuir isotherm, a separation factor (RL) proposed by Hall et al. (1966) is employed to determine whether the isotherm is favourable or not (Eq. 4). The RL values suggest whether the adsorption was favourable (0 < RL < 1), unfavourable (RL > 1), linear (RL = 1) or irreversible (RL = 0).
Freundlich isotherm is used to describe the adsorption details for heterogeneous surfaces. The isotherm model assumes that the active sites of an adsorbent have different affinities for different adsorbates (Sawyer et al. 2003). Freundlich isotherm model is expressed in Eq. 5 (Freundlich 1906).
where KF (mg/g)/(mg/L)1/n is the Freundlich constant and n is adsorption intensity. The value of 1/n determines whether the adsorption is favourable (1/n < 1) or unfavourable (1/n > 1).
The D-R isotherm is applied when the adsorbent is a porous structure. This isotherm is expressed as shown in Eq. 6 (Dubinin and Radushkevich 1947):
where qDR (mg/g) is the adsorption capacity of the adsorbent and KDR (mol2/kJ2) is a constant related to sorption energy. Polanyi potential, ε (kJ/mol), is calculated using Eq. 7.
where R (8.314 × 10–3 kJ/mol∙K) is the universal gas constant, T (K) is the temperature at which adsorption occurs, and Co (mg/g) is the concentration of the adsorbate in the standard state (1 mol/dm3) (Zhou 2020). From the D-R model, the mean adsorption energy, E (kJ/mol), can be determined as shown in Eq. 8 (Dubinin and Radushkevich 1947):
Adsorption kinetics
Adsorption kinetics were used to study the effect of contact time on the adsorption process and to identify the equilibrium time of the adsorption process (Tran et al. 2017). The experimental data was studied using four kinetic models: pseudo-first-order (PFO), pseudo-second-order (PSO), Elovich and intra-particle diffusion kinetic models. According to Ho and McKay (1998), the PFO equation is mostly appropriate for the first 20–30 min of contact time. The PFO equation proposed by Lagergren (1898) is expressed as shown in Eq. (9):
where qt (mg/g) is the amount of adsorbate adsorbed at time t (min) and k1 (1/min) is the rate constant of the PFO equation.
The PSO model, as proposed by Blanchard et al. (1984), is expressed as follows (Eq. 10):
where qt (mg/g) is the amount of Hg adsorbed at time t (min) and k2 (g/(mg × min)) is the rate constant of the PSO equation.
The Elovich equation proposed by Roginsky and Zeldovich (1934) can be expressed as follows (Eq. 11):
where α (mg/(g × min)) and β (g/mg) are the initial rate constant and the desorption constant in any one experiment, respectively.
The linearised form of the intra-particle diffusion model can be expressed as shown in Eq. 12 (Weber and Morris 1963):
where kp (mg/(g × min1/2)) is the diffusion rate constant, and C (mg/g) is the constant associated with the thickness of the boundary layer.
Results and discussion
Characterisation of AgNP
After drying the nanofibres overnight, the whitish fibre changed to a pale yellow–brown colour, indicating the formation of AgNP. SEM images were captured to observe AgNP growth on the surface of the nanofibre composites. Some backscattered electron detector SEM (BSD-SEM) images with EDS analysis of 0.01 and 0.015 M AgNP are presented in Fig. 1. The AgNP were observed on both nanofibre composites. There were relatively more distinctive nanoparticles observed on the surface of 0.01 M AgNP (Fig. 1a) than on 0.015 M AgNP (Fig. 1d). It was expected that an increase in AgNO3 concentration would contribute to either similar or relatively more AgNP on the surface of the fibre because of the higher Ag/polymer ratio in 0.015 M AgNP (Abu Bakar et al. 2007; Guidelli et al. 2011); however, SEM image proved otherwise. Even though a few AgNP were observed on the surface of 0.015 M AgNP, EDS analysis, which provides elemental composition on the surface and a few microns below the surface, showed that the AgNP were dispersed on both fibres (Fig. 1b, c, e and f). This means increasing AgNO3 concentration might have produced AgNP mostly within the fibre walls, with few on the surface in this synthesis method. Alternatively, AgNP on the outer wall of 0.015 M AgNP were probably relatively smaller, therefore being less visible in a BSD-SEM image but evident in its corresponding EDS analysis.

a BSD-SEM images with b, c EDS analysis of 0.01 M AgNP at 8106 × and d BSD-SEM images with e, f EDS analysis of 0.015 M AgNP of 0.015 M AgNP at 3932 ×
In order to verify the size of AgNP, TEM analysis was employed to image the “naked” AgNP without a polymer covering. TEM images and particle size distribution of 0.01 M AgNP and 0.015 M AgNP are presented in Fig. 2. The AgNP were virtually spherical in both nanofibres. Figure 2a and b correspond with 0.01 M AgNP at different magnifications. AgNP were well dispersed in the fibre and had a larger polydispersity with sizes from 1.57 to 19.76 nm and an average of 8.15 ± 0.09 nm (Fig. 2e). Figure 2c and d show AgNP in 0.015 M AgNP at different magnifications. Figure 2f shows that AgNP in 0.015 M AgNP had a smaller polydispersity with a size range from 0.61—19.90 nm and an average of 2.82 ± 0.04 nm. It was also observed that there were relatively more nanoparticles in 0.015 M AgNP (23.66 AgNP per 10 000 nm2 area) than in 0.01 M AgNP (2.35 AgNP per 10 000 nm2 area), which was expected due to the higher Ag/polymer in 0.015 M AgNP.

TEM images of AgNP IN 0.01 M AgNP (a and b) and 0.015 M AgNP (c and d) with their corresponding size distribution (e and f, respectively)
As observed by Guidelli et al. (2011), by comparing Fig. 2e and f, it can be seen that the size distribution of the AgNP was impacted by AgNO3 concentration. That is, higher AgNO3 concentration yielded particles with a smaller polydispersity (Fig. 2e, f). However, contrary to Guidelli et al. (2011) and Abu Bakar et al. (2007), an increase in AgNO3 concentration decreased the average size of AgNP. A study by Henglein and Giersig (1999) showed that at a lower stabilising agent/Ag ratio, the stabilising agent is not effectively built up around the Ag clusters, leading to the coalescence of Ag clusters and forming larger nanoparticles. However, in the case of 0.015 M AgNP, the matrix for the AgNP is in a solid state; therefore, there will be little room for the diffusion of Ag clusters to form larger nanoparticles. This may have accounted for its smaller dispersity. Regarding 0.01 M AgNP, since there is a relatively higher stabilising agent/Ag ratio, further reduction of the Ag ions on the surface of Ag clusters may have resulted in its larger average size (Henglein and Giersig 1999).
An XRD analysis was conducted to confirm the crystallographic structure of AgNP present in the PVA/NRL nanofibre, 0.01 M AgNP and 0.015 M AgNP. The XRD patterns for PVA/NRL and AgNP-PVA/NRL nanofibre composites are presented in Fig. 3. Due to the thin and elastomeric nature of the composites, the fibres were kept on their substrate (aluminium foil) for analysis. Characteristic peaks observed for 0.01 M and 0.015 M AgNP were at 38.3, 44.6, 65.0 and 78.2°, relative to (111), (200), (220) and (311) planes, respectively, and similar to that observed by Abu Bakar et al. (2010) and Abu Bakar et al. (2007). The peaks showed that the AgNP were face-centred cubic silver (ICDD 01–071-4612). Since aluminium peaks were intense at 44.7, 65.1 and 78.3° (ICDD 03–065-2869), as observed in PVA/NRL, these peaks caused a reduction in Ag peaks in 0.01 M and 0.015 M AgNP. However, due to the growing intensity at 38.3°, which is usually the most intense peak in Ag, it can be confirmed that the AgNP were present. The peak at 2θ = 17.0° may be attributed to an amorphous NRL halo, similar to that observed by Abu Bakar et al. (2007) but at slightly higher 2θ (20.0°).

XRD patterns for PVA/NRL and AgNP-PVA/NRL nanofiber composites
Figure 4 shows the FT-IR spectra of PVA/NRL and AgNP-PVA/NRL nanofibre composites. In the PVA/NRL nanofibre, 13 prominent peaks were observed, of which eight (2961, 2927, 2853, 1660, 1444, 1375, 1125 and 840 cm−1) were characteristic of NRL, similar to that observed by Abu Bakar et al. (2007), and four peaks (1735, 1248, 1093 and 1038 cm−1) were characteristic of PVA as observed by Bhat et al. (2005) and Mansur et al. (2008). The spectra of 0.01 M and 0.015 M AgNP were similar to the PVA/NRL spectrum; however, there was an emergence of a new peak at 1717 cm−1, and the intensities of peaks at 1735, 1248, 1125 and 1038 cm−1 significantly diminished. The diminished peak at 1735 cm−1 and appearance of 1717 cm−1 could be from a mixture of aldehydes and ketones from the carbonyl group, as observed by Abu Bakar et al. (2007). The reduced peaks at 1125 cm−1 can be attributed to CH2 wagging, and that of 1038 cm−1 can be attributed to C–N and C–O stretching. This signifies that the presence of amine group in NRL might have reduced Ag+ ions. These changes indicate an interaction between the AgNP and the PVA/NRL (Danna et al. 2016; Guidelli et al. 2011). The alterations at 759 and 738 cm−1 in 0.01 M and 0.015 M AgNP indicate that cis-isoprene may have served as a stabilising agent for the AgNP as Guidelli et al. (2011) observed. Also, as Sagitha et al. (2016) observed, the hydroxyl group in the nanofibre might have assisted in reducing Ag+ to Ago. Generally, it can be seen that both NRL and PVA played essential roles in reducing Ag+ and inhibiting AgNP agglomeration, but NRL played a major role.

FT-IR spectra of PVA/NRL nanofibre and AgNP-PVA/NRL nanofibre composites
Point of zero charge (PZC) of AgNP-PVA/NRL nanofibre composites
The results of PZC analysis are presented in Fig. 5. It was discovered that the PZC of 0.01 M and 0.015 M AgNP were 5.07 and 4.31, respectively. The PZC values aided the determination of the surface charge of nanofibre components during the adsorption of Hg.

Point of zero charge for 0.01 M and 0.015 M AgNP nanofibre composites
Effect of pH on Hg adsorption
The influence of pH on Hg adsorption by AgNP are presented in Fig. 6. The pH of a solution significantly affects the speciation of an adsorbate, which in turn impacts how adsorption occurs. In order to establish the pH at which maximum adsorption occurs, the sorption experiment was conducted at pH between 4 and 9, while contact time and initial concentration were kept constant at 60 min and 0.7 mg/L, respectively. It was found that the amount of Hg removed increased from 0.00–47.74% for 0.01 M AgNP with maximum uptake at pH 7 (45.77 mg/g), similar to the findings of Ganzagh et al. (2016). In 0.015 M AgNP, the amount of Hg removed increased from pH 4–7 (0.00–33.45%). There was a slightly higher uptake at pH 9 (23.53 mg/g) than at pH 7 (21.38 mg/g) for 0.015 M AgNP. As a result, all other experiments were conducted at pH 7. In general, 0.01 M AgNP was more efficient in Hg uptake than 0.015 M AgNP.

Effect of pH on the adsorption of Hg by AgNP nanofibre composites
Maximum Hg uptake observed at pH 7 indicates that both nanofibre composite surfaces were negatively charged at that pH. Equation (13), as suggested in Stanley L. Hem’s personal communication (Lindblad and Duroux 2017), can be used to verify this.
According to Eq. (13), the surface charges of 0.01 M AgNP and 0.015 M AgNP were −113.87 and −158.71 mV, respectively. This indicates that the nanofibre composites had a negative charge on their surfaces at pH 7. Cations are usually expected to be easily adsorbed when surfaces are negatively charged; however, at low pH, cations and hydrogen ions (H+) compete for surface sites (Drever 1997). Due to the potential abundance of H+ in solution at lower pH, H+ tends to bond directly to surface groups of adsorbents in an inner sphere complex than other cations, limiting cation adsorption (Drever 1997). This explains the limited Hg adsorption at pH below 7.
It is worth noting that at pH 7 and Eh 0.22 V (SHE), Hg exists as Hg(s) (Takeno 2005). This implies that Hg removal is not limited to cationic Hg and AgNP-PVA/NRL nanofibre composites but can also exist between metallic Hg and the nanofibre composites. At neutral pH, H+ ions balance hydroxide ions (OH−) in the solution, allowing Hg to bond freely with AgNP with minimal interference. As a result, it is likely that metallic Hg, rather than Hg2+, participated in the reaction by engulfing the AgNP on the nanofibre composite and creating an amalgam as observed by Katok et al. (2012).
Tauanov et al. (2018) and Tauanov et al. (2019) support the hypothesis that metallic Hg forms an amalgam with AgNP, as in the authors’ work, Hg2+ is reduced to Hgo as a result of a redox reaction between Ag+/Ago (+ 0.80 V) and Hg2+/Hgo (+ 0.85 V) (Sumesh et al. 2011; Henglein 1998; Henglein and Brancewicz 1997). The Hgo precipitates on the nanofibres’ surface and forms an amalgam (AgyHgz) with the AgNP in the nanocomposite, as proposed in Eqs. (14) (redox) and (15) (amalgamation) by Henglein (1998) and Henglein and Brancewicz (1997), where x represents the number of Ag clusters.
Also, according to Henglein (1998), small Ag clusters, Agx (x < 10), are electronegative. As a result, Hg2+ is reduced to Hgo while Agx is oxidised to xAg+. Considering Eq. (15), smaller values of x might prevent amalgams from forming. This explains the reason for the comparatively lower adsorption of Hg in 0.015 M AgNP, where smaller AgNP were formed. Therefore, Hg adsorption by the nanofiber composites may be attributed to many facets, including adsorption, redox reaction leading to precipitation and Hg-Ag amalgamation similar to that observed by Tauanov et al. (2018) and Tauanov et al. (2019). However, since Hg exists as quicksilver at pH 7, according to Takeno 2005, it can be said that Hg-Ag amalgamation may predominate in the removal of Hg from water.
Effect of contact time on Hg adsorption
Adsorption of Hg by the Ag-PVA/NRL nanofibre composites was studied based on contact time, as shown in Fig. 7. The pH and initial Hg concentration were kept constant at 7 and 0.5 mg/L, respectively. Since the Hg removal percentage at 0.7 mg/L was relatively low, the initial Hg concentration was slightly reduced to achieve a higher Hg removal percentage. From Fig. 7, it can be seen that PVA/NRL adsorbed Hg (35.95%); however, both AgNP nanofibre composites adsorbed a relatively higher percentage of Hg, with 0.01 M AgNP adsorbing the highest (40.92 mg/g, representing 64.30%), followed by 0.015 M AgNP (19.48 mg/g, representing 45.92%). The amount of Hg adsorbed increased with an increase in contact time up to 60 min, as observed by El-Tawil et al. (2019). After 60 min, there was a slight increase in Hg adsorption; however, it had little effect on the amount of Hg adsorbed except in 0.015 M AgNP.

Effect of contact time on the adsorption of Hg by PVA/NRL nanofibre, 0.01 M AgNP and 0.015 M AgNP
It is likely that metallic Hg easily engulfed the nanoparticles on the surface of the nanofibre composite, as observed in 0.01 M AgNP. On the other hand, the fibre walls may have retarded the diffusion rate of metallic Hg to engulf AgNP within the nanofibre, hence the lower removal efficiency of 0.015 M AgNP. The increase in Hg removal after 60 min for 0.015 M AgNP could be attributed to the concept that not all AgNP (on the surface) had been engulfed by Hg at 60 min; therefore, some active sites were still available for adsorption. Another probable reason is that after most AgNP on the fibre’s surface had been engulfed, the leftover Hg diffused into the fibre to bond with AgNP within the fibre. However, metallic Hg will diffuse at a slower rate into the bulk of the fibre than its diffusion rate from the Hg solution to the fibre’s surface. Hence, 0.01 M AgNP adsorbed Hg better, and 0.015 M AgNP was still adsorbing Hg even after 60 min. It is worth mentioning that the AgNP-PVA/NRL nanofibre composites prepared in this study were elastomeric; hence, the fibres clung to itself upon removal from the aluminium foil, which was used as a substrate during electrospinning. Therefore, the nanofibre composites could have adsorbed relatively more Hg if the fibres had not clung to itself, resulting in a reduction in its surface area for adsorption.
Effect of initial concentration on Hg adsorption
The influence of initial concentration on Hg removal from solution was studied, and the results are presented in Fig. 8. The study was conducted at an initial concentration of 0.01–1.00 mg/L, keeping pH and contact time constant at 7 and 60 min, respectively. Maximum percentage of Hg was removed at 0.05 mg/L (89.00%) for 0.01 M AgNP and 0.02 mg/L (94.50%) for 0.015 M AgNP. As the initial concentration was increased, the percentage of Hg removed decreased. It is evident that at lower Hg concentration, the ratio of Hg to adsorptive sites is lower; hence, a higher adsorption percentage is experienced. On the other hand, at a higher Hg concentration, the ratio of Hg to adsorption sites increases, resulting in more Hg competing for the same number of active sites for adsorption, hence, the lower Hg removal percentage. Notably, 0.015 M AgNP adsorbed more Hg at lower concentrations. There are more AgNP in 0.015 M AgNP, which explains why it had a higher percentage of Hg removed at a lower Hg concentration. In contrast, 0.01 M AgNP was more efficient in removing the Hg than 0.015 M AgNP at a higher Hg concentration because AgNP must have adsorbed Hg faster at the surface than diffusing into the fibre to be adsorbed by AgNP in the fibre within the 60 min time frame.

Effect of initial concentration on the adsorption of Hg by AgNP-PVA/NRL nanofibre composites
Adsorption isotherm study
The experimental data were fitted to three adsorption isotherms, namely, Langmuir, Freundlich and D-R isotherms, to describe the adsorption behaviour of Hg onto the AgNP nanofibre composites. Figure 9 illustrates the adsorption isotherms of Hg on the AgNP nanofibre composites, and Table 1 presents each model’s parameters, the corresponding coefficient of determination (R2), and the mean square errors (MSE). The experimental data of Hg removal by 0.01 M and 0.015 M AgNP were a good fit for all isotherms used in this study. For 0.01 M AgNP, it can be seen that the experimental data for Hg removal fitted best to the Langmuir isotherm with R2 = 0.9851 and MSE = 3.8067 (mg/g)2, indicating Hg adsorption by 0.01 M AgNP occurs by a monolayer adsorption process. From the isotherm, Hg adsorption by 0.01 M AgNP was found to be 68.3223 mg/g, and RL was 0.0234—0.8693. An RL value between 0 and 1 suggests that Hg adsorption was favourable. On the other hand, 0.015 M AgNP fitted best to the D-R isotherm with R2 = 0.9880 and MSE = 0.7903 (mg/g)2. This result suggests that Hg must have diffused into the pores of 0.015 M AgNP and bonded with AgNP within the fibres. The RL (0.0876–0.9057) and 1/n (0.3843) values of Hg adsorption by 0.015 M AgNP were between 0 and 1, signifying that the adsorption was favourable.

a Langmuir b Freundlich and c Dubinin-Radushkevich isotherm for Hg adsorption by AgNP-PVA/NRL nanofibre composites
By comparing Hg adsorption by 0.01 M and 0.015 M AgNP, it can be seen that 0.01 M AgNP has a higher qmax (68.3223 mg/g) than 0.015 M AgNP (26.6855 mg/g). A higher qmax signifies that 0.01 M AgNP has a better capability in adsorbing more Hg from the solution than 0.015 M AgNP. Again, by comparing the KF and qDR values of both AgNP nanofibre composites, it can be seen from Table 1 that 0.01 M AgNP (KF = 67.4295 (mg/g)/(mg/L)1/n, qDR = 962.7408 mg/g) were greater compared to 0.015 M AgNP (KF = 30.0932 (mg/g)/(mg/L)1/n, qDR = 213.1266 mg/g). Also, the smaller n value of 0.01 M AgNP (1.9716) than that of 0.015 M AgNP (2.6022) denotes better adsorption and a relatively stronger bond between the adsorbate and adsorbent in 0.01 M AgNP (Ganzagh et al. 2016). This confirms that 0.01 M AgNP was more effective in removing Hg than 0.015 M AgNP. However, the mean adsorption energy (E value) was higher in 0.015 M AgNP (15.2002 kJ/mol) than in 0.01 M AgNP (13.0690 kJ/mol). It is important to note that E values for both nanofibre composites were between the 8—16 kJ/mol range, indicating chemisorption. The better performance observed in 0.01 M AgNP may be attributed to having relatively more AgNP on the fibre’s surface than within the fibre, consequently performing better within 60 min compared to 0.015 M AgNP.
As earlier stated, the smaller adsorption capacity of 0.015 M AgNP in the 60 min time frame may likely be due to the slower rate of diffusion of the Hg from the solution into the nanofibre for adsorption by the AgNP. The values of KL followed the order 0.015 M AgNP > 0.01 M AgNP; however, the results indicate that probably not all binding sites on the 0.015 M AgNP surface may be available for Hg adsorption during the experiment’s time frame.
Adsorption kinetic study
To understand the reaction rate and sorption mechanism, the experimental data were modelled using four adsorption kinetic models: PFO, PSO, Elovich and intra-particle diffusion kinetic models. Plots of the four kinetic models are presented in Fig. 10, and their parameters are shown in Table 2. It can be seen that the experimental data fitted well to all four models but fitted best to the Elovich kinetic model for both nanofibre composites, with R2 = 0.9858 and MSE = 2.1931 (mg/g)2 for 0.01 M AgNP. The R2 and MSE values for 0.015 M AgNP were 0.9533 and 1.7198 (mg/g)2, respectively. The Elovich model is broadly used for chemisorption data, and the model provides an important parameter, β, which gives insight into the desorption of the adsorbate (McLintock 1967; Tran et al. 2017). By comparing the Elovich parameters of the two adsorbents, 0.01 M AgNP had a lower β value (0.1217 mg/g) than 0.015 M AgNP (0.3483 mg/g). For α values, 0.01 M AgNP also had a lower value (15.2885 g/(mg × min)) compared to that of 0.015 M AgNP (47.2445 g/(mg × min)). Another significant observation was that from the intra-particle diffusion model, 0.01 M AgNP had a larger C value of 9.9354 mg/g compared to 0.015 M AgNP (6.6235 mg/g).

a Pseudo-first order b pseudo-second order c Elovich and d intra-particle diffusion kinetic models for Hg adsorption by AgNP-PVA/NRL nanofibre composites
At a lower β value, it is evident that 0.01 M AgNP would have higher efficacy in removing Hg in any one experiment by desorbing a smaller amount of Hg. Despite 0.015 M AgNP having relatively more and smaller average particle size, it was expected that 0.015 M AgNP should have had a lower β value since a decrease in particle size elevates Hg adsorption, as according to the findings of Korobeinyk and Inglezakis (2018). Thus, this supports the hypothesis that in 0.015 M, though there are several AgNP, a relatively larger number exists within the fibre, and metallic Hg could not diffuse into the bulk fibre fast enough to bond with AgNP during the experiment’s time frame. The larger C value of 0.01 M AgNP, signifying a relatively larger boundary layer, indicated that though the average particle size in 0.01 M AgNP was larger, most particles were on the surface, therefore having a higher range for effective adsorption and forming stronger bonds. According to a study by Katok et al. (2012) on the interaction between Ag and Hg at the nanoscale level, the researchers observed the fastest adsorption kinetics for the smallest AgNP. Therefore, 0.015 M AgNP having a relatively smaller average size AgNP and recording a higher initial rate constant, α, was not surprising. It can be inferred that the smaller size of 0.015 M AgNP resulted in its faster initial rate of Hg adsorption from the solution to the fibre’s surface. However, as contact time increased and AgNP on the surface formed amalgams with Hg, the adsorption rate decreased due to the slower rate of Hg diffusion into the bulk fibre, hence the higher desorption constant.
Conclusion
Silver nanoparticles have been synthesised into a PVA/NRL nanofibre matrix via electrospinning and a green approach. The efficiency of the prepared nanofibre composites to remove Hg from an aqueous solution was also investigated. SEM, EDS and TEM analysis revealed that the AgNP formed were both on the surface and within the fibre, with 0.01 M AgNP (8.15 ± 0.09 nm) having a larger average size and polydispersity. The TEM analysis also showed more particles in the 0.015 M AgNP compared to 0.01 M AgNP due to a higher Ag/polymer ratio in 0.015 M AgNP. An FT-IR and XRD analysis confirmed the formation of AgNP in the fibre matrix. The percentage removal of Hg from aqueous solution by AgNP-PVA/NRL nanofibre composite depended on pH, contact time and initial Hg concentration. Maximum uptake of Hg was recorded at pH 7 for both adsorbents and around 60 min. The removal efficiency decreased with an increase in initial Hg concentration. Experimental adsorption data were fitted to Langmuir, Freundlich and D-R isotherm models. The experimental data for Hg adsorption by 0.01 M AgNP fitted best to the Langmuir isotherm, indicating monolayer adsorption. In comparison, experimental data of 0.015 M AgNP fitted best to the D-R isotherm, indicating the porous nature of the adsorbent accounted for Hg adsorption. The qmax values of the Langmuir model indicated that 0.01 M AgNP (68.3223 mg/g) was more efficient in removing Hg from aqueous solution than 0.015 M AgNP (26.6855 mg/g) within the 60 min time frame. For the adsorption kinetics, both 0.01 and 0.015 M fitted best to the Elovich kinetic model, indicating chemisorption. Lower β (0.1217 g/mg) and larger C values (9.9354 mg/g) confirmed why 0.01 M AgNP had higher efficiency in adsorbing Hg. The methodology used in this study is simple and green, raw materials are readily available, and using this nanofibre composite as an adsorbent for Hg looks promising. Additionally, because AgNP is embedded in a matrix large enough to be filtered, it can be easily removed from treated water using conventional filtration. As a result, AgNP will not remain in the treated water or require huge investments for removal following water treatment. Therefore, further investigation should be conducted to improve the synthesis and efficacy of the AgNP-PVA/NRL in removing Hg from an aqueous solution.
Data availability
All datasets generated during the current study are available from the corresponding author upon reasonable request.
References
Abu Bakar NHH, Ismail J, Abu Bakar M (2007) Synthesis and characterisation of silver nanoparticles in natural rubber. Mater Chem Phys 104:276–283. https://doi.org/10.1016/j.matchemphys.2007.03.015
Abu Bakar NHH, Ismail J, Abu Bakar M (2010) Silver nanoparticles in polyvinylpyrrolidone grafted natural rubber. React Funct Polym 70:168–174. https://doi.org/10.1016/j.reactfunctpolym.2009.11.009
Ajitha B, Reddy YAK, Reddy PS (2015) Biosynthesis of silver nanoparticles using Momordica charantia leaf broth: evaluation of their innate antimicrobial and catalytic activities. J Photochem Photobiol B Biol 146:1–9
Al-Qahtani KM (2017) Cadmium removal from aqueous solution by green synthesis zero valent silver nanoparticles with benjamina leaves extract. Egypt J Aquat Res 43:269–274
Badi’ah HI, Seedeh F, Supriyanto G, Zaidan AH (2019) Synthesis of silver nanoparticles and the development in analysis method.In: IOP conference series: earth and environmental science. 217. https://doi.org/10.1088/1755-1315/217/1/012005
Bagal MV, Raut-Jadhav S (2021) The process for the removal of micropollutants using nanomaterials. In: Bhanvase B, Sonawane S, Pawade V, Aniruddha P (eds) Handbook of nanomaterials for wastewater treatment: fundamentals and scale up issues. Elsevier, Amsterdam, pp 957–1007
Balali-Mood M, Naseri K, Tahergorabi Z, Khazdair MR, Sadeghi M (2021) Toxic mechanisms of five heavy metals: Mercury, lead, chromium, cadmium, and arsenic. Front Pharmacol 12:643972. https://doi.org/10.3389/fphar.2021.643972
Bernhoft RA (2012) Mercury toxicity and treatment: a review of the literature. J Environ Public Health 2012:460508. https://doi.org/10.1155/2012/460508
Bhat NV, Nate MM, Kurup MB, Bambole VA, Sabharwal S (2005) Effect of γ-radiation on the structure and morphology of polyvinyl alcohol films. Nucl Instrum Methods Phys Res B: Beam Interact Mater at 237:585–592. https://doi.org/10.1016/j.nimb.2005.04.058
Blanchard G, Maunaye M, Martin G (1984) Removal of heavy metals from waters by means of natural zeolites. Water Res 18:1501–1507
Danna CS, Cavalcante DGSM, Gomes AS, Kerche-Silva LE, Yoshihara E, Osorio-Roman IO, Salmazo LO, Rodriguez-Perez MA, Aroca RF, Job AE (2016) Silver nanoparticles embedded in natural rubber films: synthesis, characterisation and evaluation of in vitro toxicity. J Nanomater 2016:10
Diaz Arriaga FA (2015) Mercury in gold mining: impact in water sources intended for human consumption. Public Health Rev 16:947–957. https://doi.org/10.15446/rsap.v16n6.45406
Drever JI (1997) Adsorption. The geochemistry of natural waters: Surface and groundwater environments, 3rd edn. Prentice Hall, New Jersey, pp 87–105
Dubinin MM, Radushkevich LV (1947) The equation of the characteristic curve of activated charcoal. Proc Acad Sci USSR Phys Chem Sect 55:331–337
El-Tawil RS, El-Wakeel ST, Abdel-Ghany AE, Abuzeid HAM, Selim KA, Hashem AM (2019) Silver/quartz nanocomposite as an adsorbent for removal of mercury (ii) ions from aqueous solutions. Heliyon 5:e02415. https://doi.org/10.1016/j.heliyon.2019.e02415
Esdaile LJ, Chalker JM (2018) The mercury problem in artisanal and small-scale gold mining. Chem Eur J 24:6905–6916. https://doi.org/10.1002/chem.201704840
Freundlich H (1906) Over the adsorption in solution. J Phys Chem 57:385–471
Ganzagh MAA, Yousefpour M, Taherian Z (2016) The removal of mercury (II) from water by ag supported on nanomesoporous silica. J Chem Biol 9:127–142
Guidelli EJ, Ramos AP, Zaniquelli ME, Baffa O (2011) Green synthesis of colloidal silver nanoparticles using natural rubber latex extracted from hevea brasiliensis. Spectrochim Acta A Mol Biomol Spectrosc 82:140–145
Hall KR, Eagleton LC, Acrivos A, Vermeulen T (1966) Pore- and solid-diffusion kinetics in fixed-bed adsorption under constant-pattern condition. J Ind Eng Chem 5:212–223. https://doi.org/10.1021/i160018a011
Henglein A (1998) Colloidal silver nanoparticles: Photochemical preparation and interaction with O2, CCl4, and some metal ions. Chem Mater 10:444–450
Henglein A, Brancewicz C (1997) Absorption spectra and reactions of colloidal bimetallic nanoparticles containing mercury. Chem Mater 9:2164–2167
Henglein A, Giersig M (1999) Formation of colloidal silver nanoparticles: capping action of citrate. J Phys Chem B 103:9533–9539
Ho YS, McKay G (1998) A comparison of chemisorption kinetic models applied to pollutant removal on various sorbents. Trans IChemE 76:332–340
Jha AK, Prasad K (2010) Green synthesis of silver nanoparticles using cycas leaf. Int J Green Nanotechnol: Phys Chem 1:110–117
Katok KV, Whitby RLD, Fukuda T, Maekawa T, Bezverkhyy I, Mikhalovsky SV, Cundy AB (2012) Hyperstoichiometric interaction between silver and mercury at the nanoscale. Angew Chem 124:2686–2689. https://doi.org/10.1002/anie.201106776
Korobeinyk AV, Inglezakis VJ (2018) Silver nanoparticles synthesised within the silica matrix in hyperstoichiometrical of mercury from aqueous solutions. IOP Conference Series: Earth and Environmental Science 182: 012013. https://doi.org/10.1088/1755-1315/182/1/012013
Kumar SV, Bafana AP, Pawar P, Rahman A, Dahoumane SA, Jeffryes CS (2018) High conversion synthesis of <10 nm starch-stabilised silver nanoparticles using microwave technology. Sci Rep 8:5106
Lagergren S (1898) About the theory of so-called adsorption of soluble substances. K Sven Vetensk Handl 24:1–39
Langmuir I (1918) The adsorption of gases on plane surfaces of glass, mica and platinum. J Am Chem Soc 40:1361–1403
Li K, Jia X, Tang A, Zhu X, Meng H, Wang Y (2012) Preparation of spherical and triangular silver nanoparticles by a convenient method. Integr Ferroelectr 136:9–14. https://doi.org/10.1080/10584587.2012.686405
Lindblad EB, Duroux L (2017) Mineral adjuvants. In: Schijns V, O’hagan D (eds) Immunopotentiators in modern vaccines. Academic Press, pp 347–375
Mansur HS, Sadahira CM, Souza AN, Mansur AAP (2008) Ftir spectroscopy characterisation of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater Sci Eng C 28:539–548. https://doi.org/10.1016/j.msec.2007.10.088
McLintock I (1967) The elovich equation in chemisorption kinetics. Nature 216:1204–1205
Mohammadi F, Yousefi M, Gharemanzadeh R (2019) Green synthesis, characterisation and antimicrobial activity of silver nanoparticles (AgNPs) using leaves and stems extract of some plants. Adv J Chem A 2:266–275. https://doi.org/10.33945/sami/ajca.2019.4.1
Moldovan B, David L, Achim M, Clichici S, Filip GA (2016) A green approach to phytomediated synthesis of silver nanoparticles using Sambucus nigra L. fruits extract and their antioxidant activity. J Mol Liq 221:271–278
Mooibroek H, Cornish K (2000) Alternative sources of natural rubber. Appl Microbiol 53:355–365
Nasiruddin Khan M, Sarwar A (2012) Determination of points of zero charge of natural and treated adsorbents. Surf Rev Lett 14:461–469. https://doi.org/10.1142/s0218625x07009517
Osei LB, Ndur SA, Fosu S (2022) Synthesis and characterisation of electrospun natural rubber latex/polyvinyl alcohol for application in aqueous processes. J Rubber Res 25:313–320. https://doi.org/10.1007/s42464-022-00177-0
Park J-D, Zheng W (2012) Human exposure and health effects of inorganic and elemental mercury. J Prev Med Public Health 45:344–352. https://doi.org/10.3961/jpmph.2012.45.6.344
Roginsky S, Zeldovich YB (1934) The catalytic oxidation of carbon monoxide on manganese dioxide. Acta Phys Chem USSR 1:554
Sagitha P, Sarada K, Muraleedharan K (2016) One-pot synthesis of poly vinyl alcohol (PVA) supported silver nanoparticles and its efficiency in catalytic reduction of methylene blue. Trans Nonferrous Met Soc China 26:2693–2700. https://doi.org/10.1016/s1003-6326(16)64397-2
Salaheldin HI (2018) Optimising the synthesis conditions of silver nanoparticles using corn starch and their catalytic reduction of 4-nitrophenol. Adv Nat Sci Nanotechnol 9:10
Sawyer CN, McCarty PL, Parkin GF (2003) Chemistry for environmental engineering and science. McGraw Hill Education, New Dehli
Sumesh E, Bootharaju MS, Anshup PT (2011) A practical silver nanoparticle-based adsorbent for the removal of Hg2+ from water. J Hazard Mater 189:450–457. https://doi.org/10.1016/j.jhazmat.2011.02.061
Suwatthanarak T, Than-ardna B, Danwanichakul D, Danwanichakul P (2016) Synthesis of silver nanoparticle in skim natural rubber latex at room temperature. Mater Lett 168:31–35
Takeno N (2005) Atlas of Eh-pH diagrams: intercomparison of thermodynamic database. Geological survey of Japan open file report no.419. National Institute of Advanced Industrial Science and Technology, Research Center for Deep Geological Environments.
Tauanov Z, Tsakiridis PE, Mikhalovsky SV, Inglezakis VJ (2018) Synthetic coal fly ash-derived zeolites doped with silver nanoparticles for mercury (II) removal from water. J Environ Manag 224:164–171. https://doi.org/10.1016/j.jenvman.2018.07.049
Tauanov Z, Tsakiridis PE, Shah D, Inglezakis VJ (2019) Synthetic sodalite doped with silver nanoparticles: characterisation and mercury (II) removal from aqueous solutions. J Environ Sci Health A Tox Hazard Subst Environ Eng 54:951–959. https://doi.org/10.1080/10934529.2019.1611129
Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ (2012) Heavy metal toxicity and the environment. Exp Suppl 101:133–164. https://doi.org/10.1007/978-3-7643-8340-4_6
Tran HN, You S-J, Chao H-P (2016) Thermodynamic parameters of cadmium adsorption onto orange peel calculated from various methods: a comparison study. J Environ Chem Eng 4:2671–2682. https://doi.org/10.1016/j.jece.2016.05.009
Tran HN, You SJ, Hosseini-Bandegharaei A, Chao HP (2017) Mistakes and inconsistencies regarding adsorption of contaminants from aqueous solutions: a critical review. Water Res 120:88–116. https://doi.org/10.1016/j.watres.2017.04.014
Venkatachalam P, Geetha N, Sangeetha P, Thulaseedharan A (2013) Natural rubber producing plants: an overview. Afr J Biotechnol 12:1297–1310. https://doi.org/10.5897/AJBX12.016
Weber WJ, Morris JC (1963) Kinetics of adsorption on carbon from solution. J Sanit Eng Div 89:31–60
Zhou X (2020) Correction to the calculation of polanyi potential from Dubinnin-Rudushkevich equation. J Hazard Mater 384:121101. https://doi.org/10.1016/j.jhazmat.2019.121101
Acknowledgements
The authors are grateful to the Ghana National Petroleum Corporation (GNPC) Professorial Chair in Mining Engineering (University of Mines and Technology, Tarkwa, Ghana) for providing funds to purchase the electrospinner for this project. The authors also thank Mr David Gyapong (Esq.) for donating NRL from his farm.
Funding
The research was financially supported by the University of Mines and Technology (UMaT).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. LBO performed synthesis, characterisation and analysis and wrote the first draft of the manuscript. SF and SAN reviewed and commented on previous versions of the manuscript, and supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Editorial responsibility: S.Mirkia.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Osei, L.B., Fosu, S. & Ndur, S.A. Sorption of mercury from water by green synthesised silver nanoparticles supported on natural rubber latex/polyvinyl alcohol nanofibre. Int. J. Environ. Sci. Technol. 21, 4223–4238 (2024). https://doi.org/10.1007/s13762-023-05282-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13762-023-05282-0