
Abstract
Lennox–Gastaut syndrome (LGS) is a rare, age-related syndrome, characterized by multiple seizure types, mental regression, and specific EEG abnormalities. It is one of the most challenging epilepsy: treatment is rarely effective and the final prognosis remains poor, despite the availability of several antiepileptic drugs, validated through well-designed, randomized, controlled trials. However, it is reasonable to consider non-medical treatments, such as surgery, after failure of two-to-three drugs. This review has as goal to describe systematically the different therapeutic options for LGS, including, not only recognized antiepileptic drugs, but also new oral drugs, immune therapy, diet, surgery, and neurostimulation techniques.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lennox–Gastaut syndrome (LGS) is a rare condition classified among age-related epileptic encephalopathies. It represents 1–5% of all epilepsies and 3–10% of childhood epilepsies with an annual incidence of 0.2–2.8/10,000 births in European countries [1]. Typical age of start is between 2 and 8 years and can appear as cryptogenic (about 30%) or symptomatic of various etiologies (pre- and peri-natal insults, infection, brain tumor, and malformation including dysplasia, see Fig. 1), the latter with a worse prognosis than cryptogenic. In symptomatic LGS, it is often (about 30%) preceded by a West syndrome or focal seizures [1].

3 T FLAIR-weighted brain MRI from a 7-year-old female affected by LGS showing abnormal signal of right fronto-polar white matter consistent with a dysplasia
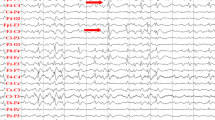
LGS is defined as an electro-clinical triad: multiple seizure types, mental regression, and specific EEG abnormalities, such as bilateral slow ( < 2.5 Hz) spike waves and fast (10–20 Hz) rhythms [2], see Fig. 2.

iEEG recording from the same patient showing generalized slow sharp-waves discharge (above) and fast rhythms originating from the right frontopolar gyrus (below)
The long-term outcome is poor and it is associated with high morbidity and mortality [3].
This review has as goal to describe systematically the different therapeutic options for LGS and summarizes all studies available on Pubmed/Medline database until the date of acceptance.
Treatment
Medical treatment
Classic antiepileptic drugs (AEDs)
Seizures in LGS are usually drug-resistant. Complete seizure control with resolution of mental dysfunction is often difficult to achieve.
Clinical trial data for classic antiepileptic drugs (AEDs) for the treatment of LGS are summarized in Table 1. Valproate, lamotrigine, and topiramate are considered the first-line drugs by many authors.
Valproate is the preferred drug for initial therapy, because it can be effective against both focal and generalized seizures. However, valproate has never been specifically licensed for use in LGS and available data for use in LGS are poor. In a first double-blind crossover trial, valproate was compared to phenobarbital and showed to be more active than phenobarbital alone in 17 LGS patients [4]. These preliminary results on valproate efficacy in LGS have been corroborated in a successive study of 336 epileptic patients, and 38 of them were LGS. About 21% of LGS patients have complete control of all seizures with valproate, and 55% experience an improvement of at least 50% in seizure frequency. Side effects were mild and included gastrointestinal effects, weight increase, transient drowsiness, hair changes, thrombocytopenia, transient lymphopenia, and abnormal liver function tests. The optimum level was between 60 and 120 mg/L, and if the daily dose does not exceed 40 mg/kg, it is free from serious adverse events [5].
Either lamotrigine or topiramate is generally the second-line choice when valproate is not effective.
Lamotrigine is authorized in Europe and the USA as adjunctive treatment in LGS. The efficacy and tolerability of lamotrigine in LGS patients were tested in a Randomized Controlled Trial (RCT) involving 169 LGS patients. Lamotrigine was started at doses of 5–50 mg/day and up-titrated to a maximum dose of 100–200 mg/day for patients receiving concomitant valproate therapy, and 300–400 mg/day for patients using lamotrigine alone. In the lamotrigine group, there was a 32% median reduction in major seizures frequency, compared to 9% in the placebo group. A reduction of all types’ seizures frequency by at least 50% was recorded in 33% of patients treated by lamotrigine than 16% of placebo group. The most common adverse events were rash, pharyngitis, fever, and infection [6].
Topiramate, as lamotrigine, is licensed in Europe and the USA as adjunctive therapy in LGS. The efficacy and tolerability in LGS were tested in an RCT including 98 LGS patients. It was up-titrated from an initial dose of 1 mg/kg/day to a maximum dose of 6 mg/kg/day. Topiramate was effective in reducing drop-attack frequency and seizure severity. The median reduction in drop-attack frequency in the topiramate group was 14.8%, compared to a 5.1% increase in the placebo group. 33% of patients had a decrease of at least 50% in major seizures with topiramate compared to 8% of patients treated by placebo. The most common adverse events were somnolence, anorexia, nervousness, behavioural problems, fatigue, dizziness, and weight loss [7].
Other effective antiepileptic drugs in LGS include levetiracetam, clobazam (and clonazepam), rufinamide, zonisamide, felbamate, and sulthiame.
Levetiracetam showed to be efficacious and well tolerated when used as an add treatment in pediatric patients with LGS. An open-label, multicenter, observational clinical trial enrolled 55 LGS patients treated with a maintenance dose of levetiracetam of 20–80 mg/kg/day. A reduction in seizure frequency of more than 50% was observed in 58.2% of patients, and 27.3% of them became seizure free. The most common adverse event was hyperactivity [8].
Clobazam is authorized in Europe as adjunctive treatment in epilepsy, and in the USA as adjunctive therapy for LGS-associated seizures. It significantly reduced the total amount of seizures and more specifically the drop seizures. The most common adverse events were upper respiratory tract infection and pyrexia [9]. Furthermore, patients receiving clobazam experienced fewer seizures related injuries than those receiving placebo [10]. Starting dose of 10–20 mg/day may be further slowly increased, even beyond the maximum indicated dose of 40 mg/day, while carefully monitoring for adverse events, with further improvement on seizure frequency [11]. Clonazepam is another benzodiazepine that may be effective in LGS, especially for myoclonic or tonic seizures, but it is associated with more common side effects compared to clobazam and has more tachyphylaxis [12, 13].
Rufinamide is approved in Europe and the USA as adjunctive therapy for LGS-associated seizures. In an RCT including 138 LGS patients, rufinamide treatment decreased drop attacks (42.5%), compared to placebo patients, with a median decrease of 32.7% in total seizure frequency compared to 11.7% for the placebo group. Doses were initiated at 10 mg/kg/day and up-titrated over 14 days to a maximum tolerated dose of 45 mg/kg/day. The most frequent adverse events were vomiting, somnolence, and rash [14].
Zonisamide was evaluated in a Korean study as adjunctive therapy in 62 LGS children. 4.8% of them were seizure free, 22.6% showed 75–100% of seizure frequency reduction and 24.2% showed 50–75% of seizure frequency reduction. It was started at an initial mean dose of 3.33 mg/kg/day and then up-titrated weekly to a mean maximum dose of 8.6 mg/kg/day. Adverse events were transient and included somnolence and anorexia [15].
Felbamate was approved in the USA and Europe as adjunctive therapy for LGS patients’ non-responders to a primary AED. An RCT involved 73 LGS patients taking an initial dose of 15 mg/kg/day felbamate and up-titrated over 14 days to a maximum dose of 45 mg/kg/day. Patients with felbamate had a decrease of 34% in frequency of atonic seizures or drop attacks, and a 19% reduction in all seizures frequency, compared to placebo with a decrease of 9% and 4%, respectively [16]. Unfortunately, felbamate carries a black-box warning for aplastic anemia and hepatic failure, which currently limits its use.
Sulthiame also showed to be efficacious and well tolerated as an add treatment in LGS. It was recently evaluated in 44 refractory LGS patients, where 61% of them had more than 50% seizure decrease. In 2%, complete seizure freedom was achieved. It was started at a dose of 100 mg/day and up-titrated over 3–8 weeks to a maximum dose of 800 mg/day. Adverse events were transient and included dyspnea, nausea, drowsiness, headache, decreased appetite, skin rash, and irritability [17].
New oral drugs
New oral drugs showed efficacy in LGS and include cannabidiol, lacosamide, perampanel, and fenfluramine. Their clinical trial data in LGS are summarized in Table 2.
Cannabis contains more than 80 phytocannabinoids, but only two molecules, tetrahydrocannabiniol and cannabidiol, have gathered the most attention based on their abundance in the plant. The potential medical use of whole-plant cannabis extracts is limited by the psychoactive properties and adverse effects associated with tetrahydrocannabinol. In the last years, enormous interest has been raised in beneficial effects in epilepsy of medical marijuana, characterized by high ratios of cannabidiol to tetrahydrocannabinol.
A multicenter open-label trial conducted on 214 patients with intractable epilepsies, 20% of them affected by LGS, provides the first prospectively collected data of cannabidiol use in children and young adults with treatment-resistant epilepsy [18]. Cannabidiol, at dose of 2–5 mg/kg/day, twice daily, was added to the baseline antiepileptic drug regimen and up-titrated by 2–5 mg/kg per week up to intolerance or 25 mg/kg/day as maximum dose. Efficacy results in LGS patients are shown in Table 2. The study also assessed interactions between other antiepileptic drugs. 51% of patients receiving clobazam had a reduction of 50% or more in motor seizures, compared to only 27% of patients not taking clobazam. Similarly, more patients taking valproate (54%) had a reduction of 50% or more in motor seizures compared to those did not taking valproate (33%) [18]. Adverse events rates were high (84–94%) [18], but most of them were deemed mild or moderate. The most common were diarrhea, somnolence, decreased appetite, pyrexia, and vomiting. Some of these adverse events may be attributable to comedication, as cannabidiol is a potent inhibitor of cytochrome P450 [19]. Thereafter, GW Pharmaceuticals has sponsored a placebo RCT for purified cannabidiol (Epidiolex) as adjunctive therapy in LGS. The cannabidiol group had a monthly frequency seizure reduction of 44% compared to 22% in the placebo group [20]. A dose-finding study was also conducted comparing cannabidiol 10 mg/kg/day, 20 mg/kg/day, and placebo, in addition to current AEDs. The mean monthly reduction in drop seizures was 17% in the placebo group compared to 42% of cannabidiol 20 mg/kg/day and 37% of cannabidiol 10 mg/kg/day [21]. The US Food and Drug Administration (FDA) approved Epidiolex in LGS for use in USA. In Europe, the European Medicines Agency (EMA) gives the Orphan Designations for Epidiolex for the treatment of LGS and some other refractory childhood-onset epilepsies.
Lacosamide is another new oral drug that showed efficacy and tolerability in LGS patients. In a multicenter retrospective study, 18 LGS children were treated by lacosamide, started as two daily doses of 1–2 mg/kg and up-titrated every 7–10 days to a mean dose of 15.2 mg/kg/day. A reduction of at least 50% in seizure frequency was noted in 33% of them. None was seizure free. The overall seizure reduction rate was 29%, in tonic seizures and drop attacks were 31% and 20%, respectively [22]. One further retrospective study conducted on 35 LGS treated by lacosamide, seizure improvement was observed in only 8.6% patients and in 5.7% by more than 50%. The highest seizure reduction was observed for focal and tonic–clonic seizures, whereas tonic and astatic seizures as well as behaviour could worsen [23]. Another study with the same design enrolled 21 children with refractory generalized epilepsy, 8 affected by LGS. In 87.5% of LGS patients, there was a seizure frequency reduction of at least 50% [24]. The most frequent adverse events were worsening of seizures, aggressiveness, irritability, weight loss, tremor, memory problems, nausea, or dizziness [22, 24].
In a prospective single-center study, 13 LGS patients were treated with perampanel as add-on therapy. A reduction of at least 50% in total seizure frequency was observed in 69.2% of patients. Improvements in cognitive function and/or behavior were reported in 53.8% of patients. The most frequent adverse events included behavior disturbance with decreased social interaction, agitation, or fatigue [25]. Other studies that included a limited number of LGS patients have showed the same results [26, 27]
A recent European study examined the effects of fenfluramine on LGS-associated seizures. The study enrolled 13 patients, given an adjunctive oral fenfluramine solution started as two daily doses of 0.2 mg/kg/day with gradual increases to a maximum of 0.8 mg/kg/day. There was a 53% median reduction in convulsive seizures and the most common adverse events were decreased appetite and decreased alertness [28].
Immunomodulatory drugs
Some small-uncontrolled studies have investigated adrenocorticotropic hormone (ACTH) and corticosteroids (such as prednisolone) in LGS.
In a study enrolling 45 LGS children who received ACTH, 51.1% of patients became seizure free for at least 10 days, and in 28.8% seizures were suppressed for over 6 months [29]. In a small trial, 86% of 7 LGS patients who received ACTH for 2 weeks showed a decrease in seizure frequency with an improvement in EEG pattern and behavior [30]. In a study of 28 epileptic children treated with prednisone for 12 weeks, 70% of the 10 LGS patients became seizure-free [31]. However, given their long-term adverse events, steroids are mainly used to treat non-convulsive status epilepticus not responding to conventional AEDs.
Small-uncontrolled trials conducted in epileptic patients receiving Intravenous Immunoglobulin (IVIG) showed evidence of improvement in both seizures and behavior. In a study of 8 LGS children, 50% showed complete remission and 37% a reduction in seizures frequency of at least 50% [32]. Two other studies, conducted on 10 and 7 LGS patients, respectively, have reported similar results [33, 34].
Ketogenic diet (KD)
The Ketogenic Diet (KD) is another effective treatment option in LGS. It should be considered early, generally in combination with pharmacological treatment. In a prospective study including 61 LGS patients, 20 patients were placed on KD and followed for a minimum of 16 months. 75% of them remained on diet for 18 months. 15% were seizure free, 15% had a 75–99% decrease in seizures frequency, 10% had a 50–74% decrease, and the remaining 35% had less than 50% decrease [35]. In a randomized blinded crossover study, 20 LGS children were enrolled. The control group was created using a glucose-containing solution, whereas the active arm received a similar-tasting saccharin solution. Over the 12-day period, 65% of patients experienced at least 50% seizure reduction. Six months after discharge, 80% had at least 50% decrease in seizures frequency, and at 12 months 65% still had at least 50% decrease [36]. Other retrospective studies, showed the same results [37, 38].
The Modified Atkins Diet (MAD) is a less restrictive alternative to the traditional KD. It is started without a fast, allows unlimited protein and fat, and does not restrict calories or fluids. A retrospective study found that it could be effective and well tolerated also in LGS children [39].
Prior to starting diet, clinic visits are strongly advised to rule out metabolic disorders and evaluate complicating comorbidities (presence of kidney stones, swallowing difficulty, hypercholesterolemia, poor weight gain or oral intake, gastroesophageal reflux, constipation, cardiomyopathy, and chronic metabolic acidosis). Screening laboratory studies should also be obtained, and include complete blood count, electrolytes, total protein, serum liver and kidney tests, lipid profile, serum acylcarnitine, vitamin D level, urinalysis, AEDs’ levels. If there is a family history of kidney stones, renal ultrasounds are advised [40]. For the follow-up, children should be seen at 1, 3, 6, 9, and 12 months on diet in the first year, then visits spaced to every 6 months, with a pediatric neurologist and dietitian seen at each visit. Discontinuation of diet should be considered after 3 months if unsuccessful, 2 years if there is benefit [40].
Surgical treatment
Patients with refractory epilepsy need to be referred to a specialized center for a presurgical evaluation, with as goal to identify seizure onset zone to potentially remove it surgically.
Surgery may consist in curative surgery of lesionectomy, lobar and multi-lobar resections, hemispheric disconnections, and palliative techniques of multiple subpial transections and callosotomy.
Curative surgery
Lesionectomy or single-lobe resection can be performed when seizure onset zone is located in one lobe or in a limited area, while multi-lobe resection may be performed when multiple epileptogenic foci are found in the same hemisphere.
Two retrospective studies of resective surgery in LGS showed promising results, particularly in patients with focal lesions. The first study was conducted on 27 LGS who underwent resective surgery. Multi-lobar resection was performed in 10 patients, single lobar resection in 11, and functional hemispherotomies in 6. No isolated lesionectomies were performed. Engel class I (seizure-free) outcomes were achieved in 59% of patients and an increase in cognitive performances in 63% [41].
The second study was conducted on 18 LGS who underwent single-lobe/lesionectomy (3 patients) or multi-lobe (15 patients) resection (plus multiple subpial transection and/or callosotomy). Results of this study showslight lower outcomes, with 40% Engel class I. The best cognitive outcome was achieved in younger patients or when the interval between seizure onset and surgery is shorter [42].
Palliative surgery
Multiple subpial transections (MST) can be a valid alternative when epileptogenic foci are located in functional areas and their removal may result in serious deficits. This technique consists in a series of transections into the cerebral cortex with the aim to interrupt some fibers that connect adjacent parts of the brain, but they do not cause long-lasting impairment in their critical functions. Complications were frequent, likely due to the direct manipulation of eloquent area. Their rate varies depending on studies. According to one study, transient paresis affects 19.8% of patients, while transient dysphasia, permanent paresis, and dysphasia 12.3%, 6.6%, and 1.9% of patients, respectively; hematoma occurs in 2.8% of patients [43]. According to another study, only 19% of patients had post-operative deficit, including memory decline, hemiparesis, and partial visual field loss [44].
Corpus callosotomy (CC) is another palliative technique that reduces frequency of generalized seizures by preventing bilateral spread of epileptic activity through the corpus callosum. This treatment is limited to patients with catastrophic intractable epilepsy and moderate to severe intellectual disability, and is considered particularly helpful for atonic, tonic, tonic–clonic seizures, and “drop attacks”. In a retrospective review of 76 LGS patients who underwent complete CC, a reduction of at least 50% in generalized seizures frequency was observed in 91% of patients, 68% had more than 90% reduction in seizure frequency, and 9% were seizure free. An increase in attention level was also observed in 86% of patients [45]. Early cases of CC encountered severe complications, such as hemispheric edema, mesial hemisphere infarcts, acute disconnection syndrome (reduction in verbal output, urinary incontinence, apathy, hemineglect), or callosal split syndrome (intermanual conflict) [1]. Recently, the classical CC has been replaced by a limited resection of the anterior region of corpus callosum, which has reduced morbidity and mortality related to CC [46]. However, complete CC is more effective than anterior CC. A prospective study examined anterior CC in 74 epileptic patients, 59 of them had LGS. A reduction in seizure frequency of more than 50% was observed in 66.2% of patients and a complete seizure-free outcome was achieved in 18.9% [47].
Neurostimulation
Vagus nerve stimulation (VNS)
In patients who are not eligible for surgery, VNS may be proposed. It is thought to be less risky than CC [48]. Side effects are relatively minimal and the more common include voice alteration, increased drooling, behavioral changes, dyspnea, and coughing. A large retrospective study reviewed VNS data from 4 Class III studies conducted in 113 LGS patients and showed that VNS is associated with a more than 50% seizure reduction in 55% of patients [49]. Another retrospective study conducted in 46 LGS patients showed the same results ( > 50% seizure reduction in 65% patients) with progressive improvement with the duration of treatment [50].
Two prospective studies, conducted, respectively, in 24 and 20 LGS patients, showed that atypical absence, myoclonic and generalized tonico-clonic seizures generally improve after VNS [51, 52]. Cognitive performance can also improve and additional positive effects include decrease in seizure severity with a quicker recovery.
Other neurostimulation techniques
Promising results in treatment of LGS were shown with the use of deep brain stimulation (DBS) and transcranial direct current stimulation (tDCS).
Deep brain stimulation (DBS) of the thalamus is an emerging treatment for medically refractory epilepsy that is not eligible for resective surgery. Among several DBS targets, thalamus centromedian nucleus (CM) stimulation is reported as a possible treatment of generalized epilepsy, in particular for LGS patients [53], although results were not significant in two studies [54, 55]. A more recent retrospective study reviewed data from 14 adult patients who underwent CM stimulation for refractory epilepsy, 4 of them with LGS. 100% of LGS patients showed a more than 50% reduction in seizure frequency [56]. The best results with CM stimulation were obtained in patients with generalized seizures, while seizures of focal origin are not significantly improved by CM stimulation in which bilateral anterior nucleus DBS is preferred [56]. This could explain why CM stimulation is more effective in LGS patients, which comprises several types of generalized seizures. The treatment is also frequently associated with an improvement in neuropsychological performance and is not complicated by neurological deficits other than hyperactive behavior, usually temporary [55, 56]
An RCT enrolled 22 LGS patients to demonstrate that tDCS, combined with pharmacologic treatment, will be more effective than pharmacologic treatment alone for reducing seizure frequency in LGS. A reduction of more than 50% in seizure frequency was observed in all individual seizure types [57]. However, further studies are needed to confirm these results.
Conclusion
LGS remains one of the most challenging epileptic encephalopathies for epileptologists, despite the availability of several new antiepileptic drugs, validated through well-designed, randomized, controlled trials. At this time, there is no evidence to suggest that any one drug is more efficacious than another, probably because published data, even from randomized studies, are difficult to compare. However, it is reasonable to consider non-medical treatments after failure of two-to-three drugs.
References
van Rijckevorsel K (2008) Treatment of Lennox–Gastaut syndrome: overview and recent findings. Neuropsychiatr Dis Treat 4(6):1001–1019
Arzimanoglou A et al (2009) Lennox–Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol 8(1):82–93
Crumrine PK (2002) Lennox–Gastaut syndrome. J Child Neurol 17(Suppl 1):S70–S75
Vassella F et al (1978) Double-blind study on the anti-convulsive effect of phenobarbital and valproate in the Lennox syndrome. Schweiz Med Wochenschr 108(19):713–716
Covanis A, Gupta AK, Jeavons PM (1982) Sodium valproate: monotherapy and polytherapy. Epilepsia 23(6):693–720
Motte J et al (1997) Lamotrigine for generalized seizures associated with the Lennox–Gastaut syndrome. Lamictal Lennox–Gastaut Study Group. N Engl J Med 337(25): 1807-1812
Sachdeo RC et al (1999) A double-blind, randomized trial of topiramate in Lennox–Gastaut syndrome. Topiramate YL Study Group. Neurology 52(9):1882–1887
Kim HJ et al (2014) Adjunctive levetiracetam treatment in pediatric Lennox–Gastaut syndrome. Pediatr Neurol 51(4):527–531
Conry JA et al (2014) Stable dosages of clobazam for Lennox–Gastaut syndrome are associated with sustained drop-seizure and total-seizure improvements over 3 years. Epilepsia 55(4):558–567
Isojarvi J et al (2016) Clobazam-treated patients with Lennox–Gastaut syndrome experienced fewer seizure-related injuries than placebo patients during trial OV-1012. Epilepsia 57(6):e113–e116
Isojarvi J et al (2018) Optimizing clobazam treatment in patients with Lennox–Gastaut syndrome. Epilepsy Behav 78:149–154
Sankar R et al (2014) Clinical considerations in transitioning patients with epilepsy from clonazepam to clobazam: a case series. J Med Case Rep 8:429
Vassella F et al (1973) Treatment of infantile spasms and Lennox–Gastaut syndrome with clonazepam (Rivotril). Epilepsia 14(2):165–175
Glauser T et al (2008) Rufinamide for generalized seizures associated with Lennox–Gastaut syndrome. Neurology 70(21):1950–1958
You SJ et al (2008) Clinical efficacy of zonisamide in Lennox–Gastaut syndrome: Korean multicentric experience. Brain Dev 30(4):287–290
Felbamate Study Group in Lennox–Gastaut, S (1993) Efficacy of felbamate in childhood epileptic encephalopathy (Lennox–Gastaut syndrome). N Engl J Med 328(1): 29–33
Caraballo RH et al (2018) Sulthiame add-on therapy in children with Lennox–Gastaut syndrome: a study of 44 patients. Seizure 62:55–58
Devinsky O et al (2016) Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol 15(3):270–278
Geffrey AL et al (2015) Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 56(8):1246–1251
GW Pharmaceutical Announces Positive Phase 3 Pivotal Study Results for Epidiolex®(cannabidiol) in the Treatment of Lennox–Gastaut Syndrome (2016) GW Pharmaceuticals website. https://www.gwpharm.com/about/news/gw-pharmaceuticals-announces-positive-phase-3-pivotal-trial-results-epidiolex. Accessed 9 Nov 2016
GW Pharmaceuticals Announces Second Positive Phase 3 Pivotal Trial for Epidiolex®(cannabidiol) in the Treatment of Lennox–Gastaut Syndrome (2016) GW Pharmaceuticals website. http://ir.gwpharm.com/news-releases/news-release-details/gw-pharmaceuticals-announces-second-positive-phase-3-pivotal-0. Accessed 9 Nov 2016
Grosso S et al (2014) Efficacy and tolerability of add-on lacosamide in children with Lennox–Gastaut syndrome. Acta Neurol Scand 129(6):420–424
Andrade-Machado R et al (2015) Efficacy and tolerability of add-on Lacosamide treatment in adults with Lennox–Gastaut syndrome: an observational study. Seizure 33:81–87
Miskin C et al (2016) Efficacy and tolerability of lacosamide in the treatment of children with refractory generalized epilepsy. J Child Neurol 31(7):925–928
Auvin S et al (2017) Use of perampanel in children and adolescents with Lennox–Gastaut Syndrome. Epilepsy Behav 74:59–63
Steinhoff BJ et al (2014) First clinical experiences with perampanel–the Kork experience in 74 patients. Epilepsia 55(Suppl 1):16–18
Huber B, Schmid G (2017) A two-year retrospective evaluation of perampanel in patients with highly drug-resistant epilepsy and cognitive impairment. Epilepsy Behav 66:74–79
Lagae L et al (2018) A pilot, open-label study of the effectiveness and tolerability of low-dose ZX008 (fenfluramine HCl) in Lennox–Gastaut syndrome. Epilepsia 59(10):1881–1888
Yamatogi Y et al (1979) Treatment of the Lennox syndrome with ACTH: a clinical and electroencephalographic study. Brain Dev 1(4):267–276
O'Regan ME, Brown JK (1998) Is ACTH a key to understanding anticonvulsant action? Dev Med Child Neurol 40(2):82–89
Sinclair DB (2003) Prednisone therapy in pediatric epilepsy. Pediatr Neurol 28(3):194–198
Gross-Tsur V et al (1993) Intravenous high-dose gammaglobulins for intractable childhood epilepsy. Acta Neurol Scand 88(3):204–209
Illum N et al (1990) Intravenous immunoglobulin: a single-blind trial in children with Lennox–Gastaut syndrome. Neuropediatrics 21(2):87–90
van Rijckevorsel-Harmant K, Delire M, Rucquoy-Ponsar M (1986) Treatment of idiopathic West and Lennox–Gastaut syndromes by intravenous administration of human polyvalent immunoglobulins. Eur Arch Psychiatry Neurol Sci 236(2):119–122
Caraballo RH et al (2014) Ketogenic diet in patients with Lennox–Gastaut syndrome. Seizure 23(9):751–755
Freeman JM et al (2009) A blinded, crossover study of the efficacy of the ketogenic diet. Epilepsia 50(2):322–325
Lemmon ME et al (2012) Efficacy of the ketogenic diet in Lennox–Gastaut syndrome: a retrospective review of one institution's experience and summary of the literature. Dev Med Child Neurol 54(5):464–468
Zhang Y et al (2016) Therapeutic effects of the ketogenic diet in children with Lennox–Gastaut syndrome. Epilepsy Res 128:176–180
Sharma S et al (2015) Use of the modified Atkins diet in Lennox Gastaut syndrome. J Child Neurol 30(5):576–579
Kossoff EH et al (2018) Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 3(2):175–192
Lee YJ et al (2010) Resective pediatric epilepsy surgery in Lennox–Gastaut syndrome. Pediatrics 125(1):e58–66
Liu SY et al (2012) Surgical treatment of patients with Lennox-Gastaut syndrome phenotype. Sci World J 2012:614263. https://doi.org/10.1100/2012/614263
Rolston JD et al (2018) Multiple subpial transections for medically refractory epilepsy: a disaggregated review of patient-level data. Neurosurgery 82(5):613–620
Spencer SS et al (2002) Multiple subpial transection for intractable partial epilepsy: an international meta-analysis. Epilepsia 43(2):141–145
Cukiert A et al (2006) Extended, one-stage callosal section for treatment of refractory secondarily generalized epilepsy in patients with Lennox–Gastaut and Lennox-like syndromes. Epilepsia 47(2):371–374
Liang S et al (2014) Anterior corpus callosotomy in school-aged children with Lennox–Gastaut syndrome: a prospective study. Eur J Paediatr Neurol 18(6):670–676
Kwan SY et al (2000) Seizure outcome after corpus callosotomy: the Taiwan experience. Childs Nerv Syst 16(2):87–92
You SJ et al (2008) Comparison of corpus callosotomy and vagus nerve stimulation in children with Lennox–Gastaut syndrome. Brain Dev 30(3):195–199
Morris GL 3rd et al (2013) Evidence-based guideline update: vagus nerve stimulation for the treatment of epilepsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 81(16):1453–1459
Cersosimo RO et al (2011) Vagus nerve stimulation: effectiveness and tolerability in 64 paediatric patients with refractory epilepsies. Epileptic Disord 13(4):382–388
Cukiert A et al (2013) A prospective long-term study on the outcome after vagus nerve stimulation at maximally tolerated current intensity in a cohort of children with refractory secondary generalized epilepsy. Neuromodulation 16(6):551–556 (discussion 556)
Cukiert A et al (2013) Long-term outcome after callosotomy or vagus nerve stimulation in consecutive prospective cohorts of children with Lennox–Gastaut or Lennox-like syndrome and non-specific MRI findings. Seizure 22(5):396–400
Li MCH, Cook MJ (2018) Deep brain stimulation for drug-resistant epilepsy. Epilepsia 59(2):273–290
Fisher RS et al (1992) Placebo-controlled pilot study of centromedian thalamic stimulation in treatment of intractable seizures. Epilepsia 33(5):841–851
Velasco F et al (2000) Predictors in the treatment of difficult-to-control seizures by electrical stimulation of the centromedian thalamic nucleus. Neurosurgery 47(2):295–304 (discussion 304-5)
Son BC et al (2016) Clinical outcome of patients with deep brain stimulation of the centromedian thalamic nucleus for refractory epilepsy and location of the active contacts. Stereotact Funct Neurosurg 94(3):187–197
Auvichayapat N et al (2016) Transcranial direct current stimulation for treatment of childhood pharmacoresistant lennox–Gastaut syndrome: a pilot study. Front Neurol 7:66
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflict of interest.
Ethical approval
This article does not include any studies on human or animal participants performed by the authors.
Informed consent
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Borrelli, S., El Tahry, R. Therapeutic approach to Lennox–Gastaut syndrome: a systematic review. Acta Neurol Belg 119, 315–324 (2019). https://doi.org/10.1007/s13760-019-01185-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-019-01185-5