
Abstract
The search for effective biological control agents without harmful non-target effects has been constrained by the use of impractical (field direct observation) or imprecise (cage experiments) methods. While advances in the DNA sequencing methods, more specifically the development of high-throughput sequencing (HTS), have been quickly incorporated in biodiversity surveys, they have been slow to be adopted to determine arthropod prey range, predation rate and food web structure, and critical information to evaluate the effectiveness and safety of a biological control agent candidate. The lack of knowledge on how HTS methods could be applied by ecological entomologists constitutes part of the problem, although the lack of expertise and the high cost of the analysis also are important limiting factors. In this review, we describe how the latest HTS methods of metabarcoding and Lazaro, a method to identify prey by mapping unassembled shotgun reads, can serve biological control research, showing both their power and limitations. We explain how they work to determine prey range and also how their data can be used to estimate predation rates and subsequently be translated into food webs of natural enemy and prey populations helping to elucidate their role in the community. We present a brief history of prey detection through molecular gut content analysis and also the attempts to develop a more precise formula to estimate predation rates, a problem that still remains. We focused on arthropods in agricultural ecosystems, but most of what is covered here can be applied to natural systems and non-arthropod biological control candidates as well.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gut content analysis has played a significant role in advancing the understanding of the feeding relationships of arthropod natural enemies, and recent advances in DNA detection place these investigations on the cusp of delineating critical details of species interactions in natural communities. In the past, prey range was done by direct field observation (live observations or video surveillance, Jones 1979; Holmes 1984; Frank et al. 2007); compilation of the scientific literature; cage or barrier experiments (exclosures or enclosures) with non-choice or choice tests (van Lenteren et al. 2006); prey baits through sentinel or artificial prey (Geiger et al. 2010); indirect inference by correlating prey and natural enemy abundance (Furlong 2015); and prey remains by visual inspection under a microscope (Ingerson-Mahar 2002). Several limitations were intrinsically related to each of those methods. Host-feeding, plant consumption (such as leaves and pollen), and predation are ephemeral events and often cryptic, especially for small organisms such as arthropods (insects and mites), so direct field observations are laborious and hard to monitor, especially for mobile natural enemies. The literature may be incomplete regarding prey and other food species of a natural enemy, especially for less studied natural enemies or those released in a new environment. Cage experiments may introduce bias that ultimately influences the natural enemy behavior and, consequently, its parasitism or predation (van Lenteren et al. 2006). In addition, they are limited to the number of trophic interactions that can be tested or predicted artificially (van Driesche and Hoddle 1997). Correlations of prey and natural enemy abundances do not demonstrate a causal predatory relation as several biotic and abiotic factors can influence their abundances (Brandon and Ives 2014). Visual analysis of prey remains under a microscope is limited to prey with hard body parts (unsuitable for fluid feeding predators), which often are not consumed by predators (Greenstone 1996), and by the degree of the sample digestion and skill of the taxonomist (Dennison and Hodkinson 1983). Rarely, in this case, does the taxonomic resolution go beyond family level.
Molecular gut content analysis is being applied to circumvent the limitations of the previous methods by analyzing prey remains in natural enemy gut contents (Symondson 2002). The evidence for predation is obtained after predation has occurred. It has been critical for assessing potential biological control efficacy (Greenstone et al. 2010; Peterson et al. 2018) and could be used to characterize environmental risks on non-target species of candidate biological control agents. The former is particularly important to target appropriate natural enemies in conservation biological control, and the latter to not harm non-target species in classical biological control. Molecular gut content analysis has also provided various estimates of predation rates (Dempster 1960; Nakamura and Nakamura 1977; Lister et al. 1987; Sopp et al. 1992; Andow and Paula, submitted), which can allow assessment of the potential impact of the natural enemy on the prey population. Finally, it is revealing the interactions of natural enemies with multiple prey species in food webs (Paula et al. 2016; Lefort et al. 2017), which may allow evaluations of impacts in a community context, and more generally, the contribution of natural enemies to community stability. In this review, we demonstrate how DNA-HTS (high-throughput sequencing) gut content analysis can be used for prey range determination, estimation of predation rates, and characterization of natural enemy food webs.
Within conservation and classical biological control, molecular gut content analysis is often overlooked. In conservation biological control, it can be used to determine the main natural enemies of key agricultural or medical pests in a habitat or ecosystem (e.g., Greenstone et al. 2010; Peterson et al. 2018). There is a tendency to rely on monitoring population fluctuations of the key pests and local natural enemies to identify the natural enemies that control the key pests. This is based on the assumption that the significant natural enemies occur in the same niche and habitat as the key pests(s) and are those with high populations when key pest(s) populations are low and vice versa. Although potentially indicative of a significant trophic interaction, the negative association of predator and prey populations does not provide sufficient cause-effect evidence that a natural enemy can control or even consume another population (Furlong 2015); not all co-occurrences represent true trophic interactions. Only direct examination of predation, such as with direct field observation and molecular gut content analysis, can accurately determine which natural enemies were frequently preying on the key pests and, therefore, which natural enemies have the potential to control the pest populations. In classical biological control, molecular gut content analysis could be used to determine the efficacy of controlling target pests and assess the potential of a natural enemy to harm non-target species.
Along with the determination of prey range, estimation of predation rates is vitally important to determine the overall effect of the natural enemy on the prey population. Ideally, predation rate is the number/biomass of prey consumed by an individual natural enemy in a specified unit of time (Dempster 1960; Hagler and Naranjo 1994). This requires that molecular gut content analysis provides quantitative information about the amount of prey consumed by a natural enemy. As will be seen below, this is controversial for some methods of molecular gut content analysis.
Most natural enemies interact with multiple species, both as consumers of prey and as the consumed prey. To fully understand their role in a community, it is important to characterize these interactions. Molecular gut content analysis can contribute to a deeper understanding of the web of trophic interactions in which a natural enemy is embedded and can begin to reveal the role of natural enemies in stabilizing community structure and possibly providing long-term suppression of prey populations. While molecular gut content analysis will not provide information on non-consumptive interactions, such as many mutualistic or trait-mediated interactions (Glossary Supplementary Information, SI), it has the power to unravel complex food webs, through determination of the many density-mediated interactions (SI Glossary) in a food web. It has contributed to the identification of considerable intraguild predation (SI Glossary) among arthropod natural enemies within an agroecosystem (Gagnon et al. 2011a, b; Davey et al. 2013; Hagler and Blackmer 2013; Raso et al. 2014; Paula et al. 2016). It was also used to verify that alternative prey are important to conserve the natural community of generalist natural enemies in the absence of pest prey species in an agroecosystem and that alternative prey can, in high abundance, disrupt predation on a pest prey (Kuusk and Ekbom 2010).
Within the molecular gut content analysis techniques, DNA high-throughput sequencing (HTS) enables analyses of a large number of samples and increases considerably the probability of detection of prey and other foods that were consumed a long time ago (days) or were rare or small. This provides a more complete prey range with a finer taxonomic resolution (Ji et al. 2013; Stein et al. 2014). This review focuses on prey range determination through gut content analysis by HTS DNA-based methods and on the construction of food webs from such data. We begin by providing a brief history of the development of molecular gut content analysis to identify prey range and trophic interactions of arthropod natural enemies. Following this, we present an overview of important issues that must be considered prior to starting DNA-based molecular gut content analysis. Then, we describe the most recent HTS molecular methods used for arthropod gut content analysis, metabarcoding and Lazaro, a method to identify prey by mapping unassembled shotgun reads, considering their strength and limitations. We then discuss how gut content analysis can be used to characterize prey range, estimate predation rates, and generate natural enemy food webs.
Prey range
Historical Overview of Molecular Gut Content Analysis
The history of molecular gut content analysis of arthropods has been described extensively (e.g., Sunderland 1988; Greenstone 1996; Symondson 2002; Harwood and Obrycki 2005; Sheppard and Harwood 2005; King et al. 2008; Furlong 2015; Birkhofer et al. 2017; Hagler 2019), so here it will be mentioned briefly. It started nearly 80 years ago using proteins as markers for the detection of prey, mostly by serological methods: precipitin test with polyclonal antibodies (Brook and Proske 1946; Hall et al. 1953; Fox and MacLellan 1956; Dempster 1960), agglutination (Greenstone 1977), complement fixation, and immunoassay (Boreham and Ohiagu 1978; Sunderland 1988). The most commonly adopted method was enzyme-linked immunosorbent assays (ELISA, polyclonal or monoclonal antibodies) (Fichter and Stephen 1981; Miller 1981; Ragsdale et al. 1981; Symondson et al. 1999, 2000; Naranjo and Hagler 2001; Hagler 2006). Non-serological methods were radio-isotope labeling (Pendleton & Grundman, 1954), paper chromatography (Putman 1965), and isoenzyme analysis (usually esterase) through electrophoresis (Murray and Solomon 1978).
Although prey detection by protein-based methods has the advantage of allowing detection of stage-specific conspecific prey, therefore enabling study of cannibalism (Sigsgaard et al. 2002), after the development of the polymerase chain reaction (PCR) in the 1980s by Mullis (1990), these methods were largely replaced by methods based on DNA (Agustí et al. 1999; Zaidi et al. 1999; Chen et al. 2000; but see Hagler 2019 on universal food immunomarking technique-UFIT). This was because the DNA-based methods were less expensive, less time-consuming and labor-intensive, and more sensitive and reproducible (Symondson 2002; Sheppard et al. 2005; although see Fournier et al. 2008). Examples of prey detection by PCR-based methods are random amplified polymorphic DNA (RAPD) (Agustí et al. 1999), microsatellite (Torr et al. 2001), PCR followed by temperature or denaturing gradient gels (TGGE and DGGE) (Harper et al. 2006; Martin et al. 2006), ligase detection reaction (LDR) PCR (Li et al. 2011), terminal restriction fragment length polymorphism (tRFLP) (Juen et al. 2012), and prey-specific primers to detect prey DNA in their natural enemy species using PCR (Zaidi et al. 1999; Agustí and Symondson, 2001; Foltan et al. 2005; Juen and Traugott 2005; Lundgren et al. 2009; King et al. 2011; Davey et al. 2013) and qPCR (Lundgren et al. 2009; Weber and Lundgren 2009).
Stable isotope analysis (15 N/14 N = δ15N and 13C/12C = δ13C; Prasifka et al., 2004; Raso et al. 2014) and fatty acid analysis (FAs) (Traugott et al. 2013) were also developed and are particularly suitable for studying seasonal changes in natural enemy diets and trophic material flows as they address linkages between natural enemies and groups of prey from different trophic levels or different basal resources. 15 N isotope accumulates up the food chain, while 13C does not, allowing the identification of the trophic level of the natural enemy (Post 2002; Layman et al. 2007). The fatty acid analysis is performed by comparing the FA profiles from the different trophic levels or groups of organisms (e.g., bacteria versus fungi), mostly for soil food webs (Ruess et al. 2002; Ferlian et al. 2012). These methods are unable to identify prey species, but can complement protein- or DNA-based methods for gut content analysis.
All these pioneering molecular methods provided important contributions for the detection of prey in predator gut contents as they demonstrated through feeding bioassays that prey detectability is influenced by several factors, including biomass of prey consumed (or meal size) (Sopp and Sunderland 1989; Hagler and Naranjo 1997; Agustí et al. 1999; Hoogendoorn and Heimpel 2001); elapsed time after prey consumption (Sopp and Sunderland 1989; Chen et al. 2000; Weber and Lundgren 2009); predator species (Sunderland et al. 1987; Sopp and Sunderland 1989; Symondson and Liddell 1993; Chen et al. 2000; Hosseini et al. 2008; Lundgren et al. 2009), stage/age (Rothschild 1966; Barazzoni et al. 2000; Cotterill et al. 2013) and sex of the predator (Symondson et al. 1999; Harwood et al. 2001); prey species (Foltan et al. 2005; Harper et al. 2005); predator feeding mode (chewing versus sucking, Greenstone et al. 2007); predator starvation period or hunger level (Lövei et al. 1985; Sunderland 1996; Weber and Lundgren 2011); subsequent food consumed (chaser diet, Weber and Lundgren 2009); temperature (Sopp and Sunderland 1989; Hosseini et al. 2008); sample preservation (Weber and Lundgren 2009); taxon-specific variation in DNA copy number per cell (in the case of comparison of quantity of different species of prey consumed (Deagle et al. 2013); primer choice (Engelbrektson et al. 2010); amplicon length (Agustí et al. 1999; Zaidi et al. 1999; Chen et al. 2000; Hoogendoorn and Heimpel 2001; Deagle et al. 2006; Hajibabaei et al. 2006; King et al. 2008), with smaller amplicons having a higher chance of detection (preferentially < 300 bp, Yu et al. 2012; although the minimum length for taxonomic discrimination should be determined, for example, Hajibabaei et al. 2006 reported it to be 109 bp for COI); and sequencing direction (Deagle et al. 2013); read quality filtering threshold (Thomas et al. 2014; Deagle et al. 2013; Nguyen et al. 2015). Later, it was demonstrated that methodological choices also affect prey detection, such as choice of metabarcode primer (Alberdi et al. 2018), sequencing platform, sequencing depth (Smith and Peay 2014), and the technique (Paula et al. 2022a). It is clear that generalizations cannot be drawn reliably from the analysis of a few samples. The lack of detection of a prey might only mean that the detection was not possible due to at least one of the aforementioned factors, as opposed to lack of predation.
These older molecular methods were used to detect specific and known prey. Prey detection and identification relied on knowing some unique particularities of the known prey, which were used to design the molecular method for that prey. For example, immunodetection methods require development of a prey species-specific antigen; PCR-based methods require identification of species-specific primers to amplify a unique DNA template sequence region; RAPD, DGGE, and SSCP require characterization of a species-specific DNA fragment length separation profile. While some of these methods can be extended to detect multiple species (e.g., multiplex-PCR, De Barba et al. 2014), they are still limited to detecting known species relying on the unique particularities of those species.
More recently, methods for determining the biodiversity of a community from environmental samples were developed that enable simultaneous identification of multiple unknown species in a sample and these methods have been applied to gut content analysis. One method relies on the concept of DNA barcoding (Hebert et al. 2003a), in which species have a DNA region with sufficient sequence divergence to resolve species taxonomy, flanked by highly conserved priming sites where “universal” primer pairs (in reality: the most general primer available, Clare 2014) could match and amplify the region for a wide range of taxa (Simon et al. 1994; Valentini et al. 2009a). A standard barcode marker for animals is a fragment of 648–658 bp of the cytochrome c oxidase I gene (COI) (Hebert et al. 2003b), known as the Folmer region (Folmer et al. 1994). This sequence is available for many species at The Barcode of Life Data System (BOLD-Ratnasingham & Hebert 2007) (http://www.boldsystems.org). The main DNA barcoding primers used to amplify various parts of the Folmer region can be found in Pompanon et al. (2012). DNA barcoding enables amplification of DNA for a wide range of taxa without prior knowledge of any part of the genome. Barcode sequences can be classified into molecular operational taxonomic units (MOTUs, Floyd et al. 2002), or if species identifications are desired, matched to a reference database of taxonomically identified barcode sequences. Barcode-based methods have been applied in gut content analysis to determine the biodiversity of prey consumed by a natural enemy, first in small scale studies through Sanger sequencing (Sanger and Coulson 1975) (Zaidi et al. 1999), and later expanded to large scale analyses with the advent of HTS. The latter is called metabarcoding and explored in more detail in a subsequent section.
Recent methods rely on analysis of DNA shotgun sequencing data without PCR amplification of the environmental samples to characterize the community structure from a DNA bulk sample taken from mock or field communities (Li et al. 2015). These methods are based on HTS of the naturally occurring multicopy genomic segments in the sample (e.g., mitochondrial DNA, plastids, and nuclear ribosomal DNA clusters), which facilitate species identifications without the need for amplification. These sequences (called reads or query sequences) are matched to one or more reference databases to identify species and have had a high rate of correct species identifications (e.g., ≥ 95%, Paula et al. 2022a). Some of these methods (e.g., mito-metagenomics, genome skimming) assemble the reads into longer contigs, which are matched to one or more reference databases (Zhou et al. 2013; Tang et al. 2014; Crampton-Platt et al. 2015; Linard et al. 2018). The longer sequence of the contigs is expected to improve species identifications. However, these assembly based methods are not suitable for gut content analysis, because the prey DNA community in the gut of a natural enemy is degraded and very difficult to assemble error free. Other methods based on DNA shotgun sequencing data are assembly free (Zhou et al. 2013; Ji et al. 2020; Sarmashghi et al. 2019; Paula et al. 2015, 2022a, b) making them suitable to be applied to gut content analysis. As these PCR-free methods preserve the original amount of DNA in the sample, they have demonstrated a positive correlation between the biomass of each identified species in the sample and the proportion of identified reads (Gómez-Rodríguez et al. 2015; Bista et al. 2018; Ji et al. 2020; Paula et al. 2022b). So far, the only PCR-free, assembly-free method demonstrated for gut content analysis of arthropod natural enemies was developed by Paula et al. (2015, 2016,2022a,b), called Lazaro, as it represents the “resuscitation” of prey identifications from degraded DNA. The application of metabarcoding and Lazaro for gut content analysis is detailed below.
DNA-HTS-Based Gut Content Analysis
Several choices and precautions need to be taken before starting any HTS gut content analysis. Most of these are guided by experience acquired from biodiversity surveys, as the use of HTS for gut content analysis in arthropods is not yet widespread. The following choices and precautions were compiled to prepare the samples to maximize detection of true positive (TP) prey and non-detection of true negative (TN) prey, and minimize detection of false-positive (FP) prey non-detection of false-negative (FN) prey. A TP is the correct detection of a prey that was consumed by the natural enemy and present in the gut contents. A FP (type I error) is the erroneous detection of a prey that in reality was not consumed by the natural enemy. These are usually biological contaminants (called “pervasive exogenous contaminants” by Taberlet et al. 2018) occurring in any step of the procedure, but for DNA-HTS gut content analysis, they can also be generated by “internal” sample artifacts, e.g., chimeras or amplification errors during PCR (Schloss et al. 2011; Taberlet et al. 2018), index jumping during library preparation and sample missassignment in the bioinformatics processing (Schnell et al., 2015), and sequencing errors (Quince et al. 2011; Taberlet et al. 2018). A TN is the correct non-detection of a species that was not consumed by the natural enemy. A FN (type II error) is the erroneous non-detection of a species that was actually consumed by the predator and present in the gut contents (Ficetola et al. 2008, 2015). Recommendations of good practices for sample preparation can be found in, e.g., King et al. (2008), Pompanon et al. (2012), Ficetola et al. (2016), and Taberlet et al. (2018). Estimation of performance measures, such as sensitivity, specificity, false discovery rate (FDR), false omission rate (FOR), and accuracy, can be found in Paula et al. (2022a). We highlight specific issues here:
-
• Decide on using non-invasive (e.g., analysis of regurgitates or feces) or invasive means (post-mortem analysis) of sample collection. Non-invasive means are preferred when one wants to release the natural enemy specimen back into nature; deposit it in an entomological collection/museum; investigate the diet of rare or protected species; track changes in individual dietary preferences over time; or not affect population densities and species interactions by taking large numbers of specimens out of a habitat (Waldner and Traugott 2012). For a non-invasive method, one will need to keep the specimen alive to collect regurgitates or feces without giving it a food supply or to collect regurgitates or feces in the wild. Regurgitation (oral fluids, crop, and/or midgut contents) is common in many arthropods as a defense mechanism, such as carabid beetles and other ground beetles (Forsythe 1982; Waldner and Traugott 2012), spiders (Kaestner 1993) and grasshoppers (Sword 2000), and it can be provoked during handling or by gently pressing anterior abdominal sternites of the predator (Forsythe 1982; Waldner and Traugott 2012). Waldner and Traugott (2012) demonstrated that the prey detection in regurgitates was similar or significantly enhanced compared to whole specimen homogenization. Advantages of regurgitates are less predator genetic material and higher relative “concentration” of prey DNA compared to invasive methods, and reduced degradation of prey DNA compared to gut contents or feces, which probably improves the accuracy of prey detection. Conversely, regurgitates only represent the most recent meal or fraction of the meal (Kamenova et al. 2018). Invasive (post-mortem) gut content analysis includes dissection of the natural enemy gut (Foltan et al. 2005) or whole body homogenization (e.g., Lundgren et al., 2009) and is usually preferred for small arthropods or ones with cryptic behavior. Most of the literature uses post-mortem analysis. In this case, the natural enemies will need to be killed and preserved immediately after collection to slow down or stop the digestion process. This can be done by immediately immersing the specimen in 70–95% ethanol followed by storage at – 20 °C or, preferably, − 80 °C (King et al. 2008). However, in our experience, adult ladybird beetles frequently regurgitate when immersed in ethanol. So, one may prefer to kill a natural enemy first by other means (e.g., killing jar with ethyl acetate) before immersing it in ethanol. Alternatively, one may extract the regurgitated DNA from the alcohol inside the collection tube. Dissection of the gut provides the advantage of reducing the ratio of predator:prey DNA compared to whole body homogenates, consequently reducing the sequencing depth required for prey detection (please see further discussion on sequencing depth for gut content analysis). Indeed, Krehenwinkel et al. (2017) recommend extracting prey DNA from the predator midgut and hindgut. However, gut dissection is difficult, time-consuming, especially for a large number of samples and/or tiny specimens (e.g., predatory mites, microhymenopteran parasitoids), and may increase the chance for DNA cross-contamination among samples.
-
• Wash potential external DNA from the natural enemy bodies. This is especially important for bulked samples, e.g., natural enemies sampled by sweeping, beating, vacuum, malaise traps, or any other way that mixes taxa (see Greenstone et al. 2012 for a recommended method to remove external DNA).
-
• Decide whether to pool or not pool specimens into a single sample (an experimental unit or replication). Pooling reduces a number of downstream steps (e.g., PCR reactions, purification, quantification, library preparation), time, and budget requirements. Pooling may be preferable when the purpose is to characterize the average diet of a population rather than individual variation. In our experience, in either case (pooling or not pooling), it is advisable to extract the DNA individually for each specimen and retain individual aliquots for follow-up work, such as confirming FPs and FNs as in Paula et al. (2022b). Generally, and whenever feasible, gut content analysis on unpooled individual specimens is better as this retains all ecological information associated with individual natural enemies and increases the number of biological replicates.
-
• Use biological replicates at the sampling step and technical replicates in the molecular methods (Taberlet et al. 2018; Ji et al. 2020). Biological replicates enable the use of ecological site occupancy models (SOMs) to estimate detection probabilities for each species, which can then be used to filter “suspicious” FP prey (Royle and Link 2006; Miller et al. 2011; Schmidt et al. 2013; Ficetola et al. 2015, 2016; Lahoz-Monfort et al. 2016). The range of biological replicates used in the literature has been 2 to 10 (Ficetola et al. 2008; De Barba et al. 2014). Technical replicates have been used in metabarcoding to filter out FP species based on the relative Euclidean distances of the number of reads (i.e., fragments of DNA sequenced) of the identified species between the PCR replicates versus among the treatments (Zinger et al. 2019; Neby et al. 2021). The higher the number of replicates, the better the prey diversity coverage (but see Smith and Peay 2014), but the higher the cost and workload. In addition, FN rates may increase if the detection probability of the TP is low (Ficetola et al. 2015). Ficetola et al. (2015) suggested a minimum 8 PCR technical replicates in metabarcoding when the probability of species detection is not high as in ancient DNA studies, and this may be the case for gut content analysis as well. Nichols et al. (2018), using rarefaction curves, verified that more than 10 PCR replicates would be necessary to sample the breath of taxa in their experiment.
-
• Plan sequencing depth of coverage. Sequencing depth of coverage is the number of times that a nucleotide position is sequenced (Sims et al. 2014). It is commonly called only “depth” or “redundancy,” often used as a synonym of “coverage,” although coverage can also refer to “breadth of coverage,” which is the proportion of genome sequenced. For genomic samples, the depth of coverage is equal to the number of reads times the average read length divided by the genome length (Sims et al. 2014). There is no standard equation for the depth of coverage for samples containing a mix of genomes or fragments of genomes, such as gut content samples and there is no widely accepted depth that recovers all taxa in such a sample. For metabarcoding, a common measure of depth is reads/amplicon/sample. Braukmann et al. (2019) constructed a mock community of 374 taxa and compared recovery of the taxa by metabarcoding in relation to coverage depth and sequencing platform (IonTorrent PGM, S5, and Illumina MiSeq). Their data show that about 80% of the taxa are recovered when there were 10,000 reads (post-filtering)/amplicon/sample and they suggested that 95% of the taxa would be recovered with 100,000–500,000 reads/amplicon/sample for all of the platforms. Although the three platforms provided similar recovery of taxa, MiSeq produced higher quality reads that facilitated bioinformatics analysis and was recommended over the other two platforms. Singer et al. (2019) conducted a meta-analysis of the 20 most indexed metabarcoding biodiversity survey studies in Google Scholar in 2018 and observed that 70% used MiSeq (probably due to lower error rates and cost compared to other technologies at the time) with a median depth of coverage of 60,000 ± 55,000 reads/amplicon/sample, ranging from 10,000 to ca. 900,000 reads. Using 8 metabarcoding samples with 3 technical replicates each, they demonstrated that increasing sequencing depth of coverage in MiSeq increased to some extent the detection rate of low-abundance taxa. But, they also demonstrated that the Illumina NovaSeq platform detected 32–40% more taxa than MiSeq when controlled for equal sequencing depth (100,000 reads/amplicon/sample) using the exact same PCR products (so no stochastic PCR biases could justify the difference in detection). Even increasing the sequencing depth, MiSeq did not reach the level of diversity detected by NovaSeq, especially for low-abundance taxa, due to its reduced capacity to detect exact sequence variants (ESVs) in samples. Moreover, taxon accumulation curves did not plateau in the NovaSeq analysis until there were 107 reads/amplicon/sample. They attributed this discrepancy in metabarcoding to the over-clustering (SI Glossary) of low diversity reads in the MiSeq flow cell, while in NovaSeq, this problem is alleviated by the patterned flow cells.
Mapping of shotgun reads has also been successfully used to characterize the biodiversity in communities. Studies with mock communities have demonstrated 94.6–97.9% accuracy in species determinations (Gómez-Rodríguez et al. 2015, Tang et al. 2015, Bista et al. 2018, Ji et al. 2020, Table S1). For these studies, we define coverage depth as the number of reads potentially hitting target genetic material times the read length divided by the average length of the target genetic material. For example, if the target genetic material is the mitogenome, then depth is the total number of reads filtered to be mitogenome reads times the read length divided by the average mitogenome size in the reference database. Gómez-Rodríguez et al. (2015) tested 10 samples containing a total of 171 chrysomelid species. Using a mitogenome reference database, they identified the species with 97.7% accuracy with an estimated depth of 3233.3. Tang et al. (2015) tested 10 bee samples containing a total of 33 species and using a mitogenome reference database they had 97.9% accuracy with an estimated depth of 268.4. Bista et al. (2018) evaluated 10 samples with 13–14 freshwater hexapods each, and using a mitogenome reference database, they had 94.6% accuracy with an estimated depth of 308.7. Ji et al. (2020) examined 14 samples with 19–20 species of Arctic arthropods each against a mitogenome reference database and had 97% accuracy with an estimated 126.8 depth.
These findings from biodiversity studies can be applied to gut content analysis by taking into account that as prey DNA is less abundant than the predator DNA in the predator gut contents and is degraded or is being degraded, the coverage depth per sample may need to be higher than for biodiversity surveys. Although there is no standard sequencing depth recommendation for gut content analysis, sufficient depth can be evaluated by rarefaction curves (Nichols et al. 2018). When targeting mitochondrial barcodes, one should consider that the proportion of mitochondrial genetic material sequenced corresponds to 0.1 to 5% of the total amount of reads sequenced (Taberlet 2012, Gómez-Rodríguez et al. 2015, Tang et al. 2015, Bista et al. 2018, Ji et al. 2020), even though mitochondrial DNA is a natural multicopy material. Also, limiting the sequencing and reducing the quantity of predator DNA will provide greater sequencing depth of prey DNA. This can be accomplished with blocking primers (Vestheim and Jarman 2008) and primers biased against the predator (Krehenwinkel et al. 2019b) for metabarcoding or dissection of the predator gut (Paula et al. 2022a) and DNA size selection for Lazaro. Some examples of sequencing depth reported in gut content analysis studies are given in Table 1. The median sequencing depth of these predator gut content studies for metabarcoding studies is 22,846 reads/amplicon/sample (range 1536 to 2,964,430 reads/amplicon/sample) and for Lazaro studies is 307.4 (range 164.4 to 803.6). For metabarcoding, this is smaller than the values used in biodiversity studies, but for Lazaro, it is a similar sequencing depth.
-
• Use a focal or comprehensive reference database. A focal reference database (e.g., Ji et al. 2020) populates the database only with the sequences of species that are expected to be found in the sample, while a comprehensive reference database (Paula et al. 2022a,b) populates the database with all available sequences of the target taxon (e.g., all arthropods, all invertebrates, all plants, or all bacterial symbionts). A focal reference database would probably reduce FPs to a minimum, because no unexpected species could be detected. However, it would also probably increase FNs, because prey could be present in the natural enemy gut that were not thought to be consumed and therefore would not be included in the reference database. Conversely, a comprehensive reference database may increase FPs, but reduce FNs. In any event, if it is not certain which species are expected to be found in the sample, a comprehensive database may be a better choice. One should be aware that reference databases typically need to be supplemented with reference sequences of local species known to be possible prey that are not already available in public sequence repositories, such as GenBank or BOLD. For example, Dopheide et al. (2019) and Liu et al. (2020) reported the lack of hundreds of OTU representative sequences in GenBank, which precluded them from identifying these species. For barcodes, this may require amplicon sequencing of positive control samples of potential prey species. For organellar genomes, this can be accomplished by including a library of each positive control sample of a potential prey species as a part of an HTS project. For all DNA-HTS methods, the taxonomic resolution of prey species identification is determined by the taxonomic resolution and accuracy of sequences in the reference database (Bridge et al. 2003). That is why it is recommended, but not always the case, to populate the reference database with sequences from morphologically curated specimens.
-
• Chose the taxonomic classifier for taxon assignment. These classifiers include alignment of the sequenced DNA using BLAST (Altschul et al. 1990), phylogeny-based methods (Munch et al. 2008a), lowest common ancestor methods (Huson et al. 2011), naïve Bayesian classifier (NBC) (Porter et al. 2014), or an alignment-free method, such as k-mer-based methods (Breitwieser et al. 2018).
Prey detection by metabarcoding
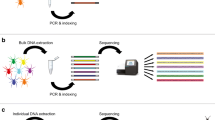
Species biodiversity assessment through DNA barcoding coupled with HTS is called “DNA metabarcoding,” first coined by Taberlet et al. (2012), but the method had already been used for species identification in environmental samples (e.g., Valentini et al. 2009a). Taberlet et al. (2018) described the method in detail in the book “Environmental DNA: For Biodiversity Research and Monitoring.” Briefly, metabarcoding starts by selecting one or more “universal” or group-specific primers to amplify a barcode region of the set of species in the sample. Considering the Illumina sequencing platform, sample DNA is amplified and tagged, preferentially with unique forward and reverse tags, during PCR amplification so that they can be pooled (multiplexed) in the same library without losing the sample identity. For library preparation, the pooled tagged samples have their ends extended by a few PCR cycles to include a library index and sequencing adaptors (sequencing priming sites and flow cell binding sites P5 and P7). Libraries are quantified (Harris et al. 2010) and, if desired, multiplexed in the same sequencing lane (usually up to 96 libraries with dual indexing sequencing) to proceed the sequencing. Depending on the desired sequencing depth per sample, the number of samples per lane can be calculated. For example, suppose one lane of an Illumina MiSeq sequencer can provide 30 million reads and you want to have at least 100,000 reads per sample and 10% of the reads will be from the calibration control library PhiX. This means that you can load up to 270 samples into the lane.
After amplicon HTS, the raw sequences pass through a quality control and then are analyzed using a bioinformatics workflow or a combination of them, available in several open source packages, initially for biodiversity surveys of microbial communities, such as MOTHUR (Schloss et al. 2009) and QIIME (Caporaso et al. 2010), later also for diet analyses, such as OBITools (Boyer et al. 2016). These packages basically will perform: (a) quality control of the dataset sequencing quality and trimming sequencing adapters; (b) reconstitute the full amplicon sequences by pairing the paired-end reads and removing non-overlapping reads; (c) sorting the amplicons from different samples to their original sample by identification of their tags (i.e., demultiplexing); clustering identical sequences and reducing them to only one, while preserving their occurrence count (dereplication); and classifying the amplicons by similarity of their sequences with thousands of sequences from the barcode region present in a reference database of several species (presumably including those from the sampled habitat) with a predefined taxonomy (supervised taxa classification) according to their similarity to a reference database; or classifying them according to their similarity to one another within a cluster, without a taxonomic reference (unsupervised classification), i.e., classifying the reads into molecular operational taxonomic units (MOTUs). In this last case, only sample richness and not sample composition is characterized. In supervised classification, taxon assignment is based on sequence similarity (match of query to the reference database) with a minimum percent identity (usually > 98%) and overlap length (usually > 100 bp) threshold, usually set arbitrarily by the researcher (Reeder and Knight 2010; Quéméré et al. 2013; De Barba et al. 2014).
Metabarcoding is a tremendous advance over previous methods for gut content analysis (e.g., Piñol et al. 2014a, b). Unlike these previous methods, it does not rely on a priori knowledge or assumptions that a species is, in fact, a prey. Most significantly, instead of screening for a single target species (prey-specific PCR, King et al. 2011; Davey et al. 2013), it screens for the entire community of prey, with an indefinite number of species that can be detected (Varennes et al. 2014). Nonetheless, the Achilles heel in metabarcoding is the use of PCR to amplify target barcode(s) and tag samples due to problems related to variation in primer efficiency of the “universal” barcode primers across a broad range of taxa (Clarke et al. 2014; Deagle et al. 2014; Elbrecht and Leese 2015, 2017), DNA polymerase errors, PCR stochasticity (mainly in the first cycles of PCR), and template switches (generation of hybrid sequences) (Kebchull and Zador 2015). Kobayashi et al. (1999) reported that errors originated in PCR amplification steps can be present in as much as 2% of all amplicons for a 250-bp amplicon.
Primer efficiency is reduced when the primer does not anneal perfectly with its template and is related to mismatches between the primer sequence and its template. In part, this happens in template protein-coding regions because of the degeneracy of the third nucleotide in a codon (Taberlet et al. 2012). Another source of mismatches is the natural variation (haplotype polymorphisms) in the nucleotide sequences (e.g., SNPs) within and between individuals from the same species or heteroplasmy, presence of different organellar genomes in the same cell or individual (Rubinoff et al. 2006). No matter the source, position, or type (Kwok et al. 1990), mismatches can result in the absence or poor amplification of the barcode for a species in the DNA mixture of a sample. Beside mismatches, Pan et al. (2014) demonstrated that primer efficiency is influenced by the DNA polymerase due to its preference for certain sequence motifs in the six nucleotides at the primer 3’-end and four nucleotides downstream of the priming site in the template DNA.
DNA polymerases errors are related to single-base substitutions, indels (insertions/deletions causing frameshift errors) (Cline et al. 1996), and errors derived from PCR stochasticity (Kebschull and Zador 2015) that lead to the formation of chimeras and heteroduplexes (Qiu et al. 2001). Chimeras may occur among closely related or abundant sequences in complex samples (Haas et al. 2011; Schloss et al. 2011; Elbrecht and Leese 2015; Taberlet et al. 2018) and are generated when incomplete extension occurs during the elongation step and the resulting fragment acts as primer in the next cycle of PCR (Quince et al. 2011). Schnell et al. (2015) discussed the different mechanisms for chimera formation and suggested that it can be avoided by using emulsion PCR as each template is amplified separately inside a microdroplet. Heteroduplexes are the result of recombination between dissimilar PCR products.
Regarding template switches, according to Pääbo et al. (1990), lesions in the template DNA, such as breaks, apurinic sites, and UV damage may cause the extending primer to “jump” to another template during the PCR. Considering this, it is reasonable to assume that the problem of template switches might happen also for gut content samples, and therefore, metabarcoding could be less appropriate for gut content analysis, as the prey DNA might be damaged due to the predator’s digestion process.
Additional potential sources of errors occurring downstream of the PCR step may arise during library construction, such as tag jumps (Amend et al. 2010; Harris et al. 2010; Porazinska et al. 2010; Carlsen et al. 2012; Schnell et al. 2015); sequencing, such as miscounted homopolymeric extensions (Kunin et al. 2010; Quince et al. 2011; Schloss et al. 2011); bioinformatic processing, such as (a) incorrect assembly of reads due to low coverage/sequencing depth (Smith and Peay 2014), (b) missorting of the reads to the correct sample (Amend et al. 2010), and (c) taxonomic overclassification (i.e., detection of a closely related species of the prey as opposed to the actual prey, which was missing from the reference database) (Richardson et al. 2017); and errors in the sequences in the reference database and mistaken taxonomy assignment of deposited sequences, both due to lack of sequence quality and taxonomic curation in most public databases (e.g., GenBank, Harris et al. 2003). Sequencing errors and taxonomic overclassification are common problems in any HTS method, not only for metabarcoding (e.g., Martin-Laurent et al. 2001; Dopheide et al. 2019).
Most of the mentioned errors can be mitigated, at least in part, by the use of algorithms specifically designed to identify and remove them (e.g., UCHIME for chimeras, Edgar et al. 2014; Edgar and Flyvbjerg 2015). If errors not removed from the datasets, they might generate FPs and/or FNs. This has created suspicion that species identified with a low number of reads are artifacts of one or more of these errors (Reeder and Knight 2010). According to Pommier et al. (2010), these potential artifacts can substantially inflate diversity estimates as they can account for more than 50% of the MOTUs after data quality control. Consequently, many authors arbitrarily remove all species identified by a small number of reads, usually < 100 reads (e.g., De Barba et al. 2014). Champlot et al. (2010), Ficetola et al. (2016), and Taberlet et al. (2018) provide guidance to avoid or minimize the occurrence of FPs and FNs. Their recommended procedures are summarized as follows:
-
A.
Employing multiple metabarcode primer pairs for the same or different barcodes (Dupuis et al. 2012; De Barba et al. 2014; Deagle et al. 2014; Krehenwinkel et al. 2017);
-
B.
Using metabarcode primers tailored for specific taxonomic groups (Harper et al. 2005; Jarman et al. 2005; Piñol et al. 2015), designed by, e.g., ecoPrimer (Riaz et al. 2011);
-
C.
Using IUPAC degenerate nucleotide codes in the metabarcode “universal” primer. While this probably reduces bias caused by mismatches of the primer with the template, it most likely also decreases primer efficiency (Jaric et al. 2013; Gibson et al. 2014);
-
D.
Using blocking primers (Vestheim and Jarman 2008) that exclude or minimize amplification of natural enemy DNA (Deagle et al. 2009; De Barba et al. 2014) or primers biased against the natural enemy (Krehenwinkel et al. 2019b), resulting in relatively more prey DNA for sequencing. This is recommended only when the natural enemy and the expected prey are phylogenetically distant, as blocking or biased primers could also block or bias the amplification of prey species closely related to the predator;
-
E.
Pretesting in vitro and/ or in silico (e.g., through ecoPCR, Ficetola et al. 2010) the metabarcode primer pairs to verify the taxonomic coverage of the expected prey and, if possible, reduce amplification of the natural enemy DNA (Clarke et al. 2014; Alberdi et al. 2018; Taberlet et al. 2018);
-
F.
Adopting good laboratory practices to minimize the risk of contamination among samples (cross-contamination) and PCR reactions (PCR product carryover) (King et al. 2008; Taberlet et al. 2018);
-
G.
Including biological and technical replicates to be able to use site occupancy models (Schmidt et al. 2013; Ficetola et a. 2015, 2016; Lahoz-Monfort et al. 2016; both in Taberlet et al. 2018) to infer the probability of detection, define a threshold to eliminate FPs, evaluate if the level of replication is appropriate to control FPs, and reduce the likelihood of FNs (Alberdi et al. 2018). For this purpose, biological and technical replicates should be sequenced separately (Smith and Peay 2014), and some advocate for sequencing separate PCR replicates for each barcode per sample (Robasky et al. 2014; Ji et al. 2020). The number of PCR replicates per sample have varied from 2 to 24 (De Barba et al. 2014; Smith and Peay 2014; Willerslev et al. 2014; Ficetola et al. 2015; Lahoz-Monfort et al. 2016; Alberdi et al. 2018; Dopheidi et al. 2019; Shirazi et al. 2021). Several studies have reported that the higher the number of PCR replicates, the higher the alpha diversity detected (specially for rarer taxa) (e.g., Alberdi et al. 2018; Dopheide et al. 2019; Shirazi et al. 2021). However, there is no general recommendation for the number of PCR replicates, as it seems to be case specific depending on the expected number of rare taxa and sequencing depth (Smith and Peay 2014; Alberdi et al. 2018). For example, for Shirazi et al. (2021), 24 PCR replicates were not enough to reach a species saturation in the rarefaction curves. Therefore, ideally a preliminary test could be conducted to estimate the number of replicates, e.g., using site occupancy models (Ficetola et a. 2015; Lahoz-Monfort et al. 2016) and rarefaction curves (Hsieh et al. 2016; Shirazi et al. 2021), both based on predictions of taxon abundance;
-
H.
Using negative DNA extraction controls, negative and positive PCR controls, unique tags and tagging system controls (Schloss et al. 2011; De Barba et al. 2014; Taberlet et al. 2018), and even a mock community (i.e., known set of organisms with quantified amounts of DNA) (Amend et al. 2010; Nguyen et al. 2015). These would enable evaluation of some sources of contamination and the efficacy of the PCR, to allow identification of tag jumps, choose objectively the appropriate sequence quality filtering threshold and calibrate the clustering threshold for MOTUs (i.e., 95%, 97%, 97.5%) to best recover the actual number of MOTUs or species in the dataset. Instead of a mock community, Shirazi et al. (2021) made a positive control PCR and library with one species known not to occur in the sampling area to be able to detect index hopping (SI Glossary) during sequencing (index hopping is also discussed in Singer et al. 2019; van der Valk et al. 2020);
-
I.
Evaluating carefully the choice of the DNA polymerase between non-proofreading versus proofreading (high fidelity) polymerases. Proofreading DNA polymerases correct for DNA amplification errors (single-base substitution errors and frameshift errors due to indels) during the elongation step in PCR because it has 3’ → 5’ exonuclease activity that removes a mismatch at the 3’-end of the new DNA strand being synthesized and replaces the incorrect nucleotide with the correct one. Sze and Schloss (2019) tested the influence of different proofreading DNA polymerases (AccuPrime, KAPA HiFi, Phusion, Platinum, and Q5) and number of PCR cycles in chimera production. They demonstrated that fewer chimera were formed using DNA polymerases with the highest fidelity and minimizing the number of PCR cycles. On the other hand, Nichols et al. (2018) evaluated two non-proofreading DNA polymerases (AmpliTaq Gold and Qiagen Multiplex Master Mix) and four proofreading DNA polymerases (KAPA HiFi, Phusion, Platinum HiFi, and Q5) to accurately estimate species occurrence and relative species abundance and the proofreading DNA polymerases did not have the best results. They reported that Platinum HiFi had a preference to amplify templates with 34–38% of GC content (polymerase GC bias) that was sufficient to distort the final result of species relative abundance. Qiagen Multiplex Master Mix had the least GC bias and, therefore, resulted in the most accurate prediction of sample species relative abundance, although it also had the highest sequence amplification errors. In addition, in complex DNA mixtures, proofreading DNA polymerases can also remove mismatches at the 3’-end of the primers, leading to non-specific amplifications (loss of specificity) (Taberlet et al. 2018) (see Taberlet et al. (2018) for a discussion of the choice of non-proofreading versus proofreading DNA polymerases);
-
J.
Optimizing PCR components and parameters to minimize PCR chimera formation, such as the use of a hotstart Taq polymerase, an appropriate number of PCR cycles (Sze and Schloss 2019), increased elongation time during library index PCR, decreased template concentration (Qiu et al. 2001; Schnell et al. 2015), and increased duration of the denaturation step (initial and in each cycle) to reduce GC bias (Aird et al. 2011). A higher number of PCR cycles might increase the likelihood of detection of rare taxa, especially in gut content analysis where the target DNA is scarce and degraded; however, it could also skew abundance estimates by amplifying the biases. Murray et al. (2015) recommend the use of qPCR to determine the optimal number of PCR cycles in the sample barcode amplification and Schnell et al. (2015) recommend the use of qPCR to determine the minimum number of PCR cycles during library preparation (tag incorporation) to minimize risk of tag jumping. Taberlet et al. (2018) recommended the use of at least 1-min elongation time to avoid the creation of artifactual sequences generated by single-stranded DNA during the downstream steps of library preparation and sequencing;
-
K.
Investing in improving the comprehensiveness, accuracy, and redundancy of the reference database for the studied ecosystem (true for any DNA-HTS-based method), preferentially with sequences from specimens in which the species had its taxonomy verified and curated. This is a special challenge for generalist natural enemies, which may have many unknown or scarce prey;
-
L.
Increasing sequencing depth per sample to reduce the possibility for FN and increase alpha diversity detected (Krehenwinkel et al. 2017; Alberdi et al. 2018; Taberlet et al. 2018). On the other hand, Shirazi et al. (2021) observed that, generally, higher sequencing depth requires a higher number of PCR replicates to reach species saturation in the rarefaction curves;
-
M.
Removing “uncertain” taxa detected only once out of the number of independent replicates (Nr) (Giguet-Covex et al. 2014; Willerslev et al. 2014; Ficetola et al. 2015).
-
N.
Calculating relative Euclidean distances of the number of reads of the identified species among the PCR replicates (not pooled) versus among the treatments (Zinger et al. 2019; Neby et al. 2021). This assumes that the distance among replicates is smaller than the distance among treatments;
-
O.
Testing species assignment using Bayesian phylogenetic analysis and provide a measure of statistical confidence in the assignment (Munch et al. 2008b);
-
P.
Filtering by minimum amplicon sequence count to remove sequences with low frequency of occurrence, either in the whole dataset or observed in a limited number of samples (Shehzad et al. 2012; Quéméré et al. 2013; De Barba et al. 2014);
-
Q.
Removing from the detection results species very unlikely to be preyed upon by a predator, either due to anatomical reasons, separation by geographic distances or seasonal patterns (De Barba et al. 2014; Taberlet et al. 2018);
-
R.
Using another method to confirm the metabarcoding results, such as melting curve analysis (MCA) in qPCR (Paula et al. 2022a).
For arthropods, the most common universal barcode primers are for the Folmer region (Folmer et al. 1994) of the COI mitochondrial gene, but many others are also used (see Valentini et al. 2009b; Pompanon et al. 2012; Taberlet et al. 2018). The COI barcode has a large reference database, but it has some important limitations, such as insufficient conservation of primer sites to allow similar primer efficiency across taxonomic groups (Deagle et al. 2014; Elbrecht et al. 2016; Sousa et al. 2019), lower taxonomic coverage than 16S (Clarke et al. 2014), and bias in amplifying lepidopterans and dipterans, while failing to amplify other insect orders (e.g., hymenopterans) (Clarke et al. 2014). A good metabarcoding primer has the combination of providing high taxonomic coverage and high taxonomic resolution, but none so far fit all the criteria (Ficetola et al. 2010; Riaz et al. 2011; Valentini et al. 2009b). One important consideration about using mitochondrial metabarcoding primers is that non-functional copies of mitochondrial genes might be transposed to the nuclear genome, creating NUMTs (nuclear mitochondrial DNA sequences). Because they lose their function (pseudogenes), they can rapidly accumulate mutations (Leite 2012) and, therefore, generate FPs.
Mapping Unassembled Shotgun Reads (Lazaro)
Lazaro, in its preliminary version called DDSS (direct DNA shotgun sequencing), is a HTS detection method developed for gut content analysis in which the prey DNA community is not enriched by PCR before sequencing and there is no read assembly after sequencing (Paula et al. 2022a). Briefly, after DNA extraction, concentrations are normalized across samples. Individual libraries are created for each sample, as there is no PCR step to tag and enable sample multiplexing in a same library, and sequenced. The raw reads are passed through quality control and, without assembly due to the degraded DNA of the prey, taxa are assigned right in the beginning of the bioinformatic workflow by matching to a single or multiple reference databases (Paula et al. 2016) using local BlastN with an E-value < 1e-30, specifying an output format XML and removing matches with overlap length < 100 bp or identity < 90% (Paula et al. 2022a). Next, a customized BlastNToSNP script is used to print the relative positions of the mismatches. Using R scripts, mismatches falsely generated by IUPAC degenerate nucleotide codes are eliminated and matches that are under a threshold of minimum percent identity (95%) and overlap length (100 bp) are discarded. A threshold is used to clean the most FPs and retain the most TPs. For 250 paired-end reads, 99% identity in 150-bp overlap was optimal (Paula et al. 2022b), and for 150 paired-end reads, 100% identity in 130 bp overlap was optimal (Paula et al. 2022a). Single-end reads are eliminated, as well as reads not mapping to coding sequence regions, e.g., of the target genome. Finally, the number of reads of species identified in the blank library are subtracted from the sample datasets. The reads of the remaining hits are then considered to identify the prey species. More details on the Lazaro methodology can be seen in Paula et al. (2022a, b) and at the GitHub repository: https://github.com/molecular-ecology/DDSS.
Lazaro shares the same advantages of metabarcoding over previous methods for DNA-based gut content analysis. It does not require a priori knowledge or assumptions that a species is a prey, and most importantly, it screens for the entire community of prey in the gut of the natural enemy (Paula et al. 2016, 2022a). However, because it does not require sample DNA amplification, it eliminates all of the limitations of metabarcoding associated with PCR, and the absence of amplification enables any part or multiple parts of the prey genome (nuclear or organellar) to be used to detect and identify prey, including any barcode sequence (Paula et al. 2016). In addition, parasites, symbionts, and plant species can also be identified in the gut contents of the natural enemy (Paula et al. 2015, 2016). These characteristics mean that the original composition of the sample DNA is preserved throughout the sequencing process and the number of reads of a prey is proportional to the amount of that prey in the original predator gut content, enabling quantitative interpretation of the results (Paula et al. 2015, 2022a, b). Moreover, this allows the samples to be reanalyzed at any time in the future when reference databases or the bioinformatic workflow have improved. Its assembly-free characteristic was designed to be suitable for applications involving degraded DNA. Instead of using assembled genomes or barcodes and sequence similarity for taxa assignment, one can use the Skmer method (Sarmashghi et al. 2019) so that unassembled reads are used in the reference database and the taxa assignment is based on counting unique k-mers. However, one should note that the absence of PCR amplification is also the source of its major limitation; samples cannot be multiplexed in the same library, because individual samples cannot be uniquely tagged. At present, this means that each sample must be made into its own library, increasing the cost of sequencing.
Some of the factors that can generate FP and FN in the species detections of metabarcoding are shared with Lazaro. For example, natural sources of mismatches (e.g., SNPs, heteroplasmy), the integrity of the DNA fragment to be sequenced, contamination in the DNA extraction, errors in the library construction, sequencing and bioinformatics steps, including taxonomic overclassification and low representativeness, and the accuracy and redundancy of species in the reference database. Based on our experience, we recommend using some of the same procedures mentioned for metabarcoding to maximize TP detection and minimize FP and FN detections with Lazaro: adopt good laboratory practices to minimize the risk of sample contamination (item F of the metabarcoding list); include biological and technical replicates (item G); use negative DNA extraction controls, a positive spike-in control community (Ji et al. 2020), and unfed predator controls (Paula et al. 2022b) (equivalent to item H); improve the comprehensiveness, accuracy and redundancy of reference databases for the studied ecosystem (item K); increase sequencing depth per sample (item L); remove “uncertain” taxa detected only once out of the number of independent replicates (item M); calculate relative Euclidean distances of the number of reads of the identified species among the replicates versus among the treatments (item N); remove from the results species very unlikely to be preyed upon by a predator (item Q); use additional method to confirm the results, as in Paula et al. (2022a) (item R).
To our knowledge, metabarcoding and mapping unassembled shotgun reads, such as Lazaro, have only been compared in two studies. Srivathsan et al. (2015) compared metabarcoding and read mapping (using BlastN) to identify diet composition by fecal analysis (host plant chloroplasts) of two red-shanked doucs langurs (Pygathrix nemaeus) fed with a known diet. While metabarcoding detected 34% of the diet composition, read mapping detected 50% of the known diet plus an unexpected species that was later confirmed to be in the diet. Paula et al. (2022a) compared metabarcoding and Lazaro prey detection in a coccinellid predator fed a mock community of prey, and in field-collected samples where detections were confirmed by qPCR-MCA. In the mock community, Lazaro detected 57% of expected prey, while metabarcoding detected none of them. In the field samples, metabarcoding and Lazaro had similar sensitivity, specificity, false discovery rate, false omission rate, and accuracy. However, prey detection was partially complementary, and while the methods shared 87% of the confirmed prey, the resulting food webs would be quite different if only one method had been used. Thus, it is not clear which, if either, of metabarcoding or Lazaro provides better prey detection in gut content analysis, but there is a slight indication that under some conditions, Lazaro may be better.
Scavenging, Secondary Predation, and Cannibalism
Even with the considerable advances in the molecular techniques for gut content analysis, the area still struggles to identify scavenging (Sunderland 1988, 1996; Calder et al. 2005; Foltan et al. 2005; Juen and Traugott 2005), secondary predation (Harwood et al. 2001; Hoogendoorn and Heimpel 2001; Sheppard et al. 2005; Paula et al. 2022b), and cannibalism (except for serological tests, Sigsgaard et al. 2002), all of which cause errors in the determination of prey range or predation rates with profound ecological implications (King et al. 2008). They are not uncommon among arthropods (they have been noted in nearly every order) and may grossly overestimate the biological control services rendered by a predator species (see several examples in Sunderland 1996). For example, intraguild predation, which can lead to secondary predation, has been observed quite commonly (e.g., Vance-Chalcraft et al. 2007; Gagnon et al. 2011a; Davey et al. 2013). An example of how secondary predation can cause a predation error is the study of Sheppard et al. (2005), who readily detected aphid DNA remains in carabid beetles that had consumed spiders (the true aphid predator). Carabid beetles, the secondary predator, had a positive detection of aphids as prey when in fact a lower trophic level predator had previously consumed the aphids. So, the actual aphid predator is not considered while the secondary predator is falsely credited with providing the biological control service.
Several attempts have been made to identify cannibalism. For example, Lövei (1986) suggested that because isoenzyme electrophoresis relies on active enzymes which may alter after death, predation could be separated from scavenging by using a combination of electrophoretic and serological tests, i.e., if serological methods detect prey but electrophoresis does not, then carrion feeding may be suspected. Sunderland (1996) proposed marking living and dead prey to study predation relative to scavenging: if living and dead prey are marked with different labels, their relative rates of consumption by predators can be studied in laboratory and field. Hagler (2019) suggested that universal food immunomarking technique (UFIT) is an ideal tool for examining arthropod scavenging and cannibalism activities. They conducted field cage studies to detect the frequency of cannibalism and intraguild predation occurring in a cotton predator assemblage. They marked early instar Chrysoperla carnea larvae with rabbit IgG, and late instars with chicken IgG. The two larval life stages (which are known to be cannibalistic) were then introduced into field cages containing other generalist predator species. The UFIT data revealed a very low frequency of cannibalism and a relatively high frequency of intraguild predation (i.e., the other generalist predators fed on the protein-marked C. carnea larvae), respectively. However, as C. carnea larvae generally do not ingest the cuticle of their prey, cannibalism may have been underestimated. A study on secondary predation by Paula et al. (2022b) using Lazaro to detect a secondarily consumed extraguild prey, Myzus persicae (Aphididae, Heteroptera), which was preyed upon first by Chrysoperla externa and secondarily by Harmonia axyridis, found that there was no significant difference in the decay rate of the M. persicae DNA as a primary or secondary prey of H. axyridis. In addition, the previous feeding history of the predator C. externa on M. persicae did not alter its DNA decay rate in the gut of H. axyridis.
Predation Rates
The previous topics addressed the challenges of accurately detecting true prey (avoiding FPs and FNs) to determine which natural enemies are preying on key pests or non-target species. For robust decision-making for biological control and risk assessment, it is necessary to go beyond prey range and determine how much predation a natural enemy may provide. Quantification of consumed prey is not easy to measure, as detected prey biomass in a predator gut is a result of meal size and time since consumption, parameters that are difficult to disentangle. In addition, it is affected by a number of aforementioned uncontrolled biotic and non-biotic factors in the field and laboratory. Estimation of predation rates is further complicated because they typically depend on the density of the prey and natural enemy. When prey are scarce, they are harder to find and predation rates are lower. When prey are highly abundant, predation rates can saturate and become independent of prey density as occurs in a type II and type III functional response (Holling 1959). When natural enemies become highly abundant, they can interfere with each other (e.g., through competition or intraguild predation), reducing the predation rate (Hassell and May 1973). These complications have yet to be addressed with molecular gut content analysis.
Quantification of prey consumption can estimate the number of prey consumed, the biomass of prey consumed, or both. The number of prey consumed is a measure of the impact of the natural enemy on the prey population, while the biomass consumed measures the impact of consumption on the natural enemy population. Sunderland (1988, 1996) argued that prey detection by molecular methods of natural enemy gut contents measures the amount of prey biomass consumed rather than the number of prey consumed. However, he pointed out that prey biomass consumed can be converted to the number of prey consumed by measuring biomass per prey and prey-size preference. He also argued that prey detection measures consumption rather than predation, because some food items can be partially consumed without killing them, and conversely, the natural enemy can kill prey but fail to ingest any prey material. Here, we use predation rates (biomass or number of prey eaten per unit of time by the entire natural enemy population) as a synonym of consumption rates (Dempster 1960; Hagler and Naranjo 1994). Some authors refer to per capita predation rates (biomass or number of prey eaten per unit of time for an individual natural enemy), such as attack rates (Lister et al. 1987). Others have used relative predator efficiency (Ragsdale et al. 1981) or predation index (Sunderland et al. 1987) as a surrogate for predation rate. There have been various terms used for the time that prey can be detected in a natural enemy gut content. Sunderland et al. (1987) clarified this by defining the maximum detectability period as the time from prey consumption to when it can no longer be detected. Implicitly, this definition is related to the limit of detection (LOD) for the detection method, and therefore, the maximum detectability period can depend on the detection method used. The terms “digestion rate,” “rate of digestion,” and “decay rate” have been used to refer to the rate at which prey decline in the natural enemy gut. We prefer “decay rate” because the rate of prey decline is related to both digestion and elimination (egestion and excretion). Greenstone et al. (2013) pointed out the difference between the decline of prey contents in a natural enemy gut (decline of analyte concentration = decay rate) and the decline in the detection of prey, such as by PCR (decline of proportion of natural enemies testing positive for prey). They proposed to call the latter a “detectability half-life,” the time when only 50% of the natural enemies test positive for prey. We will use this term to contrast with the “decay rate.” In separate work, we show that the detectability half-life is equivalent to the maximum detectability period (Andow and Paula, unpublished).
Attempts to quantify prey consumption to estimate predation rates started back when predator gut contents were analyzed by serological methods (Dempster 1960; Fichter and Stephen 1981; Sopp and Sunderland 1989; Greenstone 1996). The serological methods demonstrated that, except for Fichter and Stephen (1981), the antigen mass in the predator gut declined exponentially and the decay rate could be used to estimate biomass of ingested prey (Sopp and Sunderland 1989) and the detectability period of the biomass. Models employing the Poisson distribution (Nakamura and Nakamura 1977; Lister et al. 1987) were efforts to estimate predation rates from the frequency of predation. Several of the early works for estimating predation rates from gut content analysis in arthropods were based on a combination of predator density, proportion of predators positive for prey detection, and detection period of the prey remains in the predator gut determined experimentally in feeding trials.
In the pioneering work of Dempster (1960), predation rate was related to the proportion of the predator population (p) that fed on the prey (in his case, the chrysomelid beetle Phytodecta olivacea Forster, detected by the precipitin test) within a certain number of hours. He sampled 11,286 arthropod predators from 19 taxa to estimate predation rates (k) for each predator species. He first determined the detection period (which he called rate of digestion) for each predator (about 1 day) and estimated the likelihood of a predator preying on multiple prey during 1 day by observing the mobility of the predators in an insectary and the mean distance between the prey eggs and larvae. He argued that predators were highly unlikely to prey on multiple prey during a day because prey were far apart compared to the mobility of the predators and estimated the daily rate of predation for each predator in the community with k = (P × p)/D, where P is the predator density, and D is detection period. However, his predation rate equation was criticized because it was only applicable to predators consuming a single prey during the prey detectability period, which was considered unusual, except when the prey is bigger than the predator or rare. This predation rate equation was considered, in most cases, to underestimate predation rates.
Rothschild (1966), studying predation rates on Conomelus anceps (Germar) (Homoptera: Delphacidae) by 91 predator species by precipitin tests (only 29 species tested positive), adjusted Dempster’s (1960) equation by introducing what he called the rate of feeding (kl), i.e., average number of prey consumed during the detectability period (D), measured in the laboratory. So k = (P × p × kl)/D (in Dempster 1960kl = 1). Later, Kuperstein (1979) estimated predation rates on Eurygaster integriceps Puton (Hemiptera: Scutelleridae) by five species of carabids and independently developed the same equation for predation rate as Rothschild (1966).
Nakamura and Nakamura (1977) studied predation on the small chestnut gall wasp Dryocosmus kuriphilus Yasumatsu (Hymenoptera: Cynipidae) by precipitin test in numerous taxa of spiders (1063 individuals). They assumed a random distribution of the number of prey consumed over time and, therefore, the distribution of the number of prey consumed would be Poisson. It is well known that the zero term of the Poisson distribution is equal to the value p × kl in Rothschild (1966), so k = P × [ln(1-p)]/D, where in their case D was 4 days. Their equation was criticized because it can lead to overestimated predation rates if the predators feed on a few large prey or have a long detection period, although neither criticism applied to their case.
Ragsdale et al. (1981) assessed predation by 11 predators of the soybean pest Nezara viridula (L.) (Hemiptera: Pentatomidae) using ELISA. They used only relative predator densities and the proportion of the predator population giving a positive detection to generate a surrogate predation rate: relative predator efficiency = [(P × p)/∑(P × p)] × 100. They did not include the rate of consumption of prey by the various predators or the detection period of the prey antigen in their equation.
In 1983, Hance and Rossignol (cited by Sunderland 1988) used ELISA to quantify predation by Bembidion quadrimaculatum (L.) (Coleoptera: Carabidae) on Megoura viciae (Kaltenbach, 1843) (Hemiptera: Aphididae). They proposed that the effects of the meal size (M) and time since feeding (t) could be separated by a regression of a dilution series of the predator gut content (x) on the absorbance reading (y) as they thought absorbance reading at the y-axis intercept was related only to the meal size. However, in a subsequent work, they did not observe the same relationship and could not separate M and t (Sunderland 1988).
Sunderland et al. (1985), cited in Sunderland 1988) proposed a method to improve estimates of predation rates. They suggested that any method that quantified biomass in a predator could be applied to field-collected predators. They suggested measuring this over a period of days and taking an average over those days (they called this average x/y). They then proposed that predation rate could be estimated by k = (Px)/(yzD), where z is a constant that adjusts the effect of D on the predation rate. They suggested that D needed to be adjusted by z because prey biomass declines exponentially with time in the predator. They further proposed that z = 0.5 may be a reasonable value.
Sunderland et al. (1987) examined which of several polyphagous predators (1,275 individuals from the field distributed in Diptera, Carabidae, Coleoptera, Linyphiidae, Staphylinidae, Dermaptera, etc.) were preying on the cereal aphids Sitobion avenae (F.), Metopolophium dirhodum (Wlk.), and Rhopalosiphum padi (L.) and which had higher predation indexes using quantitative data from ELISA. They combined the percentage of predators positive for prey detection (p) and predator density (P) with laboratory estimated detectability periods. They were the first to use the terminology Dmax for detectability period, i.e., the maximum period over which prey antigens could no longer be detected in the gut of any predator individuals in feeding trials in the laboratory. Predation indexes were obtained by the equation: Predation index = (P × p/Dmax). In this work, they demonstrated that variations in Dmax of a prey among predators can be significant, so they suggested that Dmax should always be estimated to compare which predators were controlling aphid populations as a longer detectability period could lead to an overestimate of the predation rate by that natural enemy. Greenstone et al. (2010) agreed that variation in the detectability period should be incorporated in estimates of predation rate, but suggested an alternative to Dmax, which they called the detectability half-life. They reported large variation in the detectability half-life of a single egg of the prey Leptinotarsa decemlineata (Say) (Coleoptera: Chrysomelidae) by conventional PCR. In larval Coleomegilla maculata (DeGeer) (Coleoptera: Coccinellidae), it was only 7.0 h while in nymphal Perillus bioculatus (Fabricius) (Hemiptera: Pentatomidae) it was 84.4 h. Providing a correction of the predation rate with the detectability half-life or detectability period can be used to rank the potential of predator species as biological control agents as done in several other studies (e.g., Chen et al. 2000; Hosseini et al. 2008; Gagnon et al. 2011b).
Lister et al. (1987) estimated predation rates using two methods. They studied the amount of predation on Cryptopygus antarcticus Willem (Collembola: Entomobryidae) and several other prey groups by the predaceous mite Gamasellus racovitzai (Trouessart) (Mesostigmata: Ologamasidae) (about 2000 individuals from 14 localities in Antarctica, quantitative electrophoresis). They quantified the amount of prey in a predator by measuring the content of an esterase band (quantified by scanning transmission densitometry using green light 510 nm on an RFT Transidyne 2955 scanning densitometer, in arbitrary units) in an isoenzyme electrophoresis profile. They noted that a proportion, b of the predators did not search for prey, and generalized the Nakamura and Nakamura (1977) method to take this into account to estimate predation rates. Here we convert their formula using the previously defined parameters, including their b, to estimate biomass of prey eaten for the predator population: k = P × (1–b) × -ln[(1–p–b)/(1–b)]/D. More importantly, they were the first to provide a method to calculate predation rates from quantitative data of prey biomass in predator gut contents using decay rate instead of detectability period. They showed that Qj(t), the quantity of esterase stain in band j declined exponentially with time, t, after prey consumption, and specifically Qj(t) = bj M exp(-djt), where t is the time after ingestion of a meal of size M (measured in μg), bj is a constant that converts μg ingested prey into electrophoresis band units, and dj (> 0) characterizes the decay rate of the band. The quantity of prey consumed was calculated as the difference in live weight of the predator before and after being allowed to feed on the prey for 12 h. The predation rate, using previously defined parameters is k = P × (dj × p × Qj*)/(bj × M), where M is the mean size of meal ingested, Qj* is the average quantity of prey in predators with positive traces of prey measured in electrophoresis band units and Qj*/(bj × M) is the proportion of initial prey biomass remaining in the predator.
Sopp and Sunderland (1989) used quantitative ELISA to detect the aphid Sitobion avenae (F.), in the guts of 12 predator species in the Linyphiidae, Carabidae, and Staphylinidae in feeding trial experiments. The absorbance values of positive wells were converted to prey biomass (mg) using a calibration curve. The mean consumed prey biomass was then estimated for each predator species and time interval at each temperature. To enable direct comparison between predator species and temperatures in the rate of antigen decay, the biomass values for subsequent time intervals were divided by those for immediately after feeding (t = 0) to give the proportion of the biomass present at t = 0 remaining at each subsequent time interval. They showed that the size of the meal has a positive relation with the prey absorbance, the detection period did not depend very much on the initial meal size, and that the decay rate did not depend on the amount of prey initially consumed. Moreover, in 35 of 36 experiments, decay was exponential. They did not estimate predation rates. These results supported a similar finding by Fichter and Stephens (1984), also using ELISA, for an insignificant effect of meal size on the detection period.
In 1992, Sopp et al. published another method to estimate predation rates using prey biomass detected by quantitative ELISA (and a calibration curve) in the predator gut. The predation rates of biomass of prey (mg/day) were calculated from the formula: k = P × Q0 /(f × Dmax), where Q0 is the average quantity of prey measured in the predator gut content (including those with no prey detected), f is a constant related to the way the prey biomass decays over time and is equal to the average amount of prey left in the gut of a random predator at the end of the detection period. They studied predation on the cereal aphid Sitobion avenae (F.) by Agonum dorsale (Pont.) and Bembidion lampros (Herbst) (Coleoptera: Carabidae), Tachyporus hypnorum (F.) (Coleoptera: Staphylinidae), and Erigone atra Blackwell (Araneae: Linyphiidae). They compared the predation rates obtained by their equation with the observed predation rates in laboratory and insectary experiments, and compared its performance with the predation rate estimates using the methods proposed by Dempster (1960), Rothschild (1966), Kuperstein (1979), and Nakamura and Nakamura (1977). They found that predation rates based on their method were close to those measured in the laboratory experiments, while all of the other methods underestimated predation rates.
Greenstone and Hunt (1993) introduced the concept of detectability half-life and the importance of determining it, as opposed to Dmax, to enable more accurate comparison of predation rates among predators with different digestion rates. Later, Chen et al. (2000) provided a simple, robust method for estimating the detectability half-life using probit regression, and demonstrated that the detectability half-life can be significantly different for different predator species feeding on the same prey species. They advocated for the determination of detectability half-life to adjust estimated predation rates to enable “fair” comparisons of predation rates among different predator species preying on the same prey species. They advocated the use of detectability half-life as a correction factor (e.g., Greenstone et al. 2010; Hosseini et al. 2008; Gagnon et al. 2011b) as opposed to Dmax (Sunderland et al. 1987) for the cases where prey DNA detectability decays exponentially during digestion.
Naranjo and Hagler (2001) built on these methods and attempted to add a functional response to prey density to enable estimation of predation for varying prey densities. They studied predation by Geocoris punctipes (Say) (Hemiptera: Geocoridae) and Orius insidiosus (Say) (Hemiptera: Anthocoridae) on eggs of Pectinophora gossypiella (Saunders) (Lepidoptera: Gelichiidae) in greenhouse cages using qualitative ELISA. They used an empirical functional response model based O’Neil and Stimac (1988):
where Na is the predation rate (number of prey attacked by the predator population density P), p is the proportion of predators testing positive for the prey, N is the prey density, D is the detectability period, which may depend on temperature, θ, and the parameters C1, C2, and C3 describe details of the searching behavior of a natural enemy (the interested reader is referred to O’Neil and Stimac 1988). Using an independent experiment, they compared their functional response equation to the Dempster (1960) and Nakamura and Nakamura (1977) predation rate equations. They found that their equation fit the observed data better than the others. This is not surprising as their model uses 4 additional parameters than the others, which use only 3 parameters to fit the data. Overall, Naranjo and Hagler’s (2001) result suggests that the functional response needs to be considered when estimating predation rates.
Attempts have been made to quantify arthropod predation rates using quantitative PCR (qPCR) (Zhang et al. 2007; Durbin et al. 2008; Lundgren et al. 2009; Weber and Lundgreen 2009; Lundgren and Fergen 2011). Zhang et al. (2007) estimated the number of the red-eyed Bemisia tabaci (Homoptera: Aleyrodidae) B-biotype preyed upon by several field-collected predators (coccinellids Propylaea japonica, Harmonia axyridis, and Scymnus hoffmanni, the chrysopids Chrysopa pallens and C. formosa, the hemipteran Orius sauteri, and the spiders Erigonnidium graminicolum and Neoscona doenitzi) with absolute quantification by qPCR. This involves creating a standard curve to relate Cq values (quantification cycle: the cycle number at which the fluorescence signal intercepts the baseline threshold) to the quantity of B. tabaci DNA and using the standard curve to convert sample Cq values to DNA quantity. Following best practices, the standard curve was amplified in the same qPCR run as the samples. This is an absolute estimate of the quantity of B. tabaci DNA in the predator gut, as opposed to the relative estimates described above.
Lundgren et al. (2009) and Lundgren and Fergen (2011) used the Cq values obtained in a qPCR to estimate relative predation indices of consumption of western corn rootworm Diabrotica virgifera (Coleoptera: Chrysomelidae) by the natural enemy in the community. The relative prey consumption index = P × p × (100/Cq), where 100/Cq is an index of the amount of prey DNA in the natural enemies. While they were correct to say that there is a negative correlation between the concentration of prey DNA in a sample and the Cq, the relationship between them is highly non-linear, so the index is not directly related to predation rates. Although not with arthropods, Deagle and Tollit (2007) used qPCR to test if they could estimate the relative quantity of DNA of three prey fish species (50%, 36% and 14% by mass in the food) in the feces of captive sea lions. They observed that, although the absolute amount of prey DNA varied considerably, the percent composition of fish mtDNA roughly corresponded to the mass of fish in the food mixture (57.5 ± 9.3%, 19.3 ± 6.6%, and 23.2 ± 12.2%, respectively). They inferred that there was variation in the digestion rate of the different prey species in the predator gut.
The previous methods to estimate predation rates typically aimed to quantify predation on only one or a few prey species. More challenging is the simultaneous quantification of the biomass of prey consumed for the diversity of prey present in a predator gut to estimate predation rates on multiple prey. Metabarcoding and Lazaro are promising methods for doing this using relative read abundance (RRA) as a proxy for species quantity in a predator gut; however, they are presently potentially limited by some biological and technical factors that influence RRA, as mentioned previously section, including taxon-specific variation in DNA copy number per cell (in the case of comparison of quantity of different species of prey consumed) and the read quality filtering threshold (Thomas et al. 2014; Deagle et al. 2013; Nguyen et al. 2015).
Metabarcoding has been used to estimate species relative biomass or numerical abundance in two ways: by counting the frequency of occurrence of each detected species in a set of samples or by relative read abundances (e.g., Kowalczyk et al. 2011; Rayé et al. 2011; Thomas et al. 2014; Deagle et al. 2009; Soininen et al. 2009; Murray et al. 2011; Brown et al. 2012; Pinto & Raskin 2012; Deagle et al. 2019). The first is uncontroversial and provides the same information as used by Dempster (1960) and others mentioned above. The second is based on an expectation that metabarcoding RRA would correlate positively with prey biomass in predator guts. Relative differences in RRA has been used to quantify two closely related plant species in sheep diets (Willerslev et al. 2014), the taxonomic composition of mammalian herbivore diets (Kartzinel et al. 2015) and bacterial gut communities combined with digital PCR (dPCR) (Barlow et al. 2020) at the family level, and the eDNA of fish populations (Di Muri et al. 2020). Although these might be special cases, they provide hope that metabarcoding RRA can be related quantitatively to prey biomass in predator guts. As metabarcoding relies on PCR and read assembly, RRA is prone to have quantification biases related to variation in species-specific primer efficiency, amplicon length (shorter amplicons may artificially increase species richness and evenness), primer tag jumps, and barcode primers preference to amplify certain taxonomic groups (Amend et al. 2010; Engelbrektson et al. 2010; Berry et al. 2011; Ihrmark et al. 2012; Pinto & Raskin 2012; Deagle et al. 2013, 2014; Clarke et al. 2014; Elbrecht and Leese 2015; Alberdi et al. 2018), which leads to lack of or misrepresentation of taxa or over-/underestimation of interaction strength (Yu et al. 2012; Leray et al. 2013; Deagle et al. 2014; Elbrecht and Leese 2015; Piñol et al. 2015; Bista et al. 2018; Lamb et al. 2018; Piñol et al. 2018; but see Willerslev et al. 2014; Kartzinel et al. 2015; Thomas et al. 2016; Krehenwinkel et al. 2017). Perhaps the most critical bias for quantification is species-specific variation in primer efficiency. For example, suppose two prey were equally abundant in the sample, but prey 1 had an amplification efficiency of 2.0, the theoretical maximum, and prey 2 had an amplification efficiency of 1.9. After 25 amplification cycles, prey 1 would have 3.6 times more read than prey 2. Thus, it would be a mistake to conclude that there was more prey 1 than prey 2 in the original sample. As a second example, suppose prey 1 was 1/10 the abundance of prey 2, but its amplification efficiency was 2.0 and prey 2 had an acceptable efficiency of 1.8. After 25 cycles, prey 1 would have 1.4 more reads than prey 2, and it would be a serious mistake to conclude that prey 1 was more abundant than prey 2 in the original sample. Therefore, there remains controversy about the use of metabarcoding RRA for quantitative analysis (Valentini et al. 2009a; Piñol et al. 2015, 2018; Thomas et al. 2016; Bista et al. 2018; Deagle et al. 2019; Lamb et al. 2018), i.e., to estimate host preference, predation rates, or population-level interaction strengths (Deagle et al. 2019; Lamb et al. 2018).
To control for the many biases mentioned previously, Thomas et al. (2014) proposed the use of sequencing in parallel “control materials” (similar to “spike-in standards”) with a diet composition matched to the sample diets to generate correction factors for the RRAs related to species-specific biases originating from biological differences in DNA extraction, PCR amplification, and sequencing. They called these correction factors “tissue correction factors” (TCFs), which are calculated as the proportion of a prey in the control material mixture divided by the proportion of reads of that prey in the amplicon pool of the control material mixture. They applied these TCFs to a diet of known composition of three fish species consumed by harbor seals determined from scat samples. The use of TCFs reduced the average difference between the prey proportions in the diet provided to the seals and the proportion of reads of the respective prey from 28 to 14%. However, they did not produce the same rank order of prey as in the scat samples. Later, Thomas et al. (2016), recognizing the impracticality of matching the control material to the diet of a field-collected individual, proposed using a series of relative correction factors (50/50 RCFs) to correct RRAs, which are calculated from 50/50 mixtures of a control prey species and a target prey species. However, they found that RCFs were not constant and were strongly dependent on the proportional composition of the mixture of control and target species. In the worst case observed, RCF varied from 0.4 to 2.0 as the proportion of the target species increased from 20 to 80%. The methods of Thomas et al. (2014, 2016) have also been criticized as they seem applicable only for a single environmental sample or replicates from the same location involving phylogenetically similar prey taxa. Lamb et al. (2018) conducted a meta-analysis of metabarcoding RRAs and found a significant, but weak, relation between the input material for each species present and the proportions of reads and suggested the inclusion of mock communities to facilitate the assessment of the quantitative metrics. Murray et al. (2011) compared the performance of qPCR and HTS (454 GS-Junior) to quantify the relative amount of fish present in the fecal material of the little penguin Eudyptula minor and found that the proportional composition of the four most abundant food were correlated. However, they had only an average of 290 reads per sample, which greatly limited primer bias, and the use of proportions eliminates variation related to the relative amounts of food consumed, i.e., RRA. Together, however, these studies suggest that it may become possible to use RRA from metabarcoding to quantify prey biomass in predator gut contents.
Quantification of multiple prey using Lazaro RRAs has shown promise. Paula et al. (2022b) tested it to quantify the amount of prey, the aphid Myzus persicae, consumed by the coccinellid predator Hippodamia convergens and found that the number of prey reads detected was directly and quantitatively related to the number of prey consumed and time since consumption (r2 = 0.932). Moreover, the number of prey consumed did not influence the prey DNA decay rate in the predator gut. The higher the number of prey consumed, the longer was its Dmax in the predator guts. They demonstrated how to predict the number or biomass of prey consumed and how long ago consumption occurred for predator samples from the field using an inverse regression, when the number of prey reads and prey DNA decay rates are known. While it might be expected that digestion would be slower in a full gut (Weber & Lundgren 2009), their result suggests that the rate of decay of prey reads was not significantly affected by the amount of prey consumed. Paula et al. (2022a) also showed that the number of prey reads was correlated with the probability of prey detection by melting curve analysis (MCA) and that the number of reads was proportional to the relative template concentration measured by qPCR, i.e., proportional to the amount of prey DNA in the predator gut. These are promising results enabling quantitative interpretation of RRA using Lazaro, however, additional studies are needed in a variety of ecological contexts before quantitative interpretation of RRA can be confidently done.
We present in the following the information necessary to quantify per capita predation rates on a prey. To facilitate this discussion, we use two parameters that have been measured by molecular gut content analysis: p, the proportion or frequency of natural enemies that test positive for a prey, and Q*, the quantity of the prey detected in a natural enemy given that prey was detected (i.e., this excludes the individuals without prey). Consequently, Q = pQ* is the average prey quantity in a natural enemy, where Q includes individuals without prey). If pQ* can be assumed to be in a steady state, i.e., staying about the same over time, then pQ* is the balance between intake of prey DNA via predation (per capita predation rate, kpc) and the loss of prey DNA from the natural enemy by decay (which includes digestion and excretion, d). Andow and Paula (unpublished) show that per capita predation rate is
and predation rate (by the natural enemy population, P) is
Thus, estimating the decay rate, d, of Q is essential for estimating a predation rate. A high predation rate or a low decay rate can generate a high Q, and conversely, a low predation rate or a high decay rate can generate a low Q.
To evaluate the impact of the natural enemy on the prey population, it is necessary to estimate the predation rate per prey, N, and per natural enemy, P. This is because impact is estimated using population models, the simplest of which is the Lotka-Volterra predator–prey model (which is related to the Nicholson-Bailey parasitoid-host model). The Lotka-Volterra model is
where kpc is the per capita predation rate of the natural enemy P, r is the intrinsic rate of increase of the prey population N, c is a coefficient converting prey into natural enemy offspring, and γ is the natural enemy death rate. For discrete populations, it is
where k = kpcP is the predation rate as defined in this paper.
Most of the molecular gut content methods estimate k = kpcP as the impact of the natural enemy on the prey. In the Lotka-Volterra model, it can be seen that kpcP is a good estimate of the impact of the natural enemy on the prey. However, this simple Lotka-Volterra model is considered too unrealistic to estimate natural enemy impact on a prey, because it assumes that predation rate is a constant proportion of available prey. Specifically, it assumes that predation is a type I functional response, which means that the natural enemy never becomes saturated. Elaborations of the Lotka-Volterra model allowing a non-linear predator functional response are considered more realistic for evaluating the impact of the natural enemy on a prey. Nearly all functional response models take the form
where Na is the number (density) of prey attacked, N is the number (density) of prey available, \({k}_{pc}^{^{\prime}}\) is the per capita predation rate by a natural enemy when prey are scarce, and g(N) is a function describing the non-linear effect of prey number (density) on the predation rate. This formulation of the functional response allows the Lotka-Volterra model to be elaborated as follows
and
Thus, it can be seen that the method used by most molecular gut content analysis (kpcP) estimates the linear part of the functional response, and a challenge that remains is to estimate the non-linear part, specifically, \({k}_{pc}^{^{\prime}}g\left(N\right)\), instead of just kpc. This requires estimation of per capita predation, kpc, at several prey densities and fitting the non-linear function to the result. This has only been attempted by Naranjo and Hagler (2001) as discussed in detail above. In any case, predation rates determined by molecular gut content analysis alone are not a sufficient indicator to rank natural enemies for pest suppression. Other contemporaneous prey mortality methods should also be used as an additional metrics of predation, for example, predation on sentinel prey (Lundgren and Fergen 2011).
Food Webs
Predation rates alone are not a sufficient indicator to rank natural enemies for pest suppression. This is because natural enemies interact with each other as competitors and intraguild predators, and interact with other non-pest prey species and plants as omnivores, and pollen and nectar feeders. These interactions are needed to better understand the pest suppression potential of natural enemies and their potential to adversely affect non-target species. Food webs are considered to be essential for understanding the dynamics of these complex interactions. A food web is a network of populations connected by their trophic interactions. Food webs can be used to evaluate how predation on a prey may resonate through the community, which has profound implications for biological control of pests and risk assessment analysis of non-target species. To our knowledge, there are only two publications using DNA-HTS for food web construction in the context of biological control. Paula et al. (2016) using Lazaro were able to identify several direct and indirect species interactions and trophic levels, including intraguild predation, in the ladybird beetle community in a brassica agroecosystem. Lefort et al. (2017) using metabarcoding of aphid mummies were able to identify the aphid species and the parasitoids and hyperparasitoids associated with the mummies and compare the structure of the food webs in the native and invaded range of the aphid.
There are different kinds of food webs (e.g., community food webs, energy flow webs, functional food webs, Cohen 1978, Dunne 2006), but the one that DNA-HTS gut content analyses are eminently suitable for is the construction of consumer centric food webs (so-called sink food webs), which begin by identifying the consumed prey. An alternative approach is resource centric food webs (so-called source food webs, Cohen 1978). These focus on a particular food resource and identify all the consumers feeding on the resource and the consumers of those consumers (e.g., Memmott et al. 2000; Muller et al. 1999). As gut content analysis does not reveal all consumers of a resource, source food webs will not be considered further. Here, we address how sink food webs can be constructed from molecular gut content analyses and explore how they can be used to address questions of indirect interactions and community stability as well as new questions associated with network analysis.
Food webs comprise populations (nodes) and trophic linkages, which are lines connecting prey and consumer populations. Food webs can either be qualitative or quantitative. In a qualitative food web, the trophic relations between predator and prey are indicated by presence or absence and they are more common because the data requirements are much less than for a quantitative web. However, only limited inferences can be drawn from qualitative webs. For example, rare feeding events are given equal weight as common events, information about relative food preferences is not included and communities are more likely to be considered unstable.
In a quantitative food web, the trophic linkages are quantified. There are several ways to quantify (Berlow et al. 2004), but we consider two that are relevant to molecular gut content analysis: the probability of prey consumption (p) and the amount of prey consumed per predator (Q). We do not address the estimation of population dynamic interaction strengths here, such as per capita change in prey population growth rate, as these require additional information on the change in prey population densities over time. These trophic linkages are per capita effects of an individual predator on the prey. The probability of prey consumption is estimated from the proportion or frequency of natural enemies that test positive for the prey, p, which is a per capita estimate. The biomass of prey consumed per predator is estimated from d × p × Q*, as detailed previously. Both the probability and the amount will likely vary with the prey and predator densities, as measured by the functional responses. Thus, while a quantitative food web provides considerably more information than a qualitative web, it is considerably more challenging to construct and will be contingent on the existing community composition.
Once the feeding linkages have been characterized either qualitatively or quantitatively, they can be assembled into an adjacency matrix (for qualitative linkages) or a flow matrix (for quantitative linkages) (SI Glossary). These matrices can be used to graph the food web and analyze food web structure, as detailed below. An adjacency matrix comprises 0’s (predation not confirmed, i.e., prey not detected) and 1’s (predation confirmed, i.e., prey detection), while a flow matrix comprises real numbers or functions ≥ 0. Both matrices are square matrices, meaning that they have the same number of rows and columns. Each population is arrayed in the columns and appears in the same order in the rows. When in a row, the population is the prey, and when in a column it is the predator. This means that any column i and row i indicates the consumption of the ith population by itself, i.e., cannibalism. Table 2 indicates feeding relations between some hypothetical predators and prey, and Fig. S1 is the associated adjacency matrix. A graph of this food web can be constructed using igraph in R (Figs. 1 and S2) showing the trophic level of each population (TL, SI Glossary) and the trophic linkages among populations.

Graph of a qualitative food web, plotted using igraph in R. Lines are linkages and arrowheads indicate the direction of consumption or the flux of energy. Circles are nodes and represent populations (orange are basal species that do not consume other populations in the food web; blue are consumers). Height on the y-dimension is proportional to the trophic level. Trophic level for basal populations is set to 0. The trophic level of consumers is the average of all the prey consumed by a population plus 1. For example, Pop7 consumes Pop5 and Pop6, which are both trophic level 0. The average trophic level of Pop5 and Pop6 is 0, so the trophic level of Pop7 is 1. Pop 1 consumes Pop5 and Pop7, which have a trophic level of 0 and 1. Thus, the trophic level of Pop1 is 1.5. Pop3 is cannibalistic and therefore has an arrow that loops to itself. Its trophic level is 1.5 and involves mathematically complex calculations (see trophic level in SI Glossary). Pop4 is a top predator of the food web because it is not consumed by any other population. Its trophic level is determined from the average of the trophic levels of Pop1 (1.5), Pop7 (1), and Pop2 (0), and is 1.83
Community stability is an important property that can be evaluated using food webs. One significant form of stability is that all populations in the community can persist without going extinct, i.e., community richness is stable. This would imply that any non-target effects of a natural enemy would not cause the loss of a non-target species. A stronger, more restricted form of stability is that all populations will tend to some long-term steady state. This means that the population densities will tend to go to some value specific to that population. These values would allow estimation of the biocontrol service of a natural enemy as well as its impact on non-target species. Here we examine some properties of food webs that appear to be associated with community stability.
Elton (1927) hypothesized that natural enemies play a key role in stabilizing animal food webs in both senses of stability. He suggested that because they switch prey to feed on the most common prey, they will tend to keep herbivore populations in check. This idea lent support to the broader diversity-stability hypothesis that greater diversity resulted in more stable communities. Theoretical studies, however, showed that greater diversity would result in less stable communities (Levins 1970; May 1972), and present-day researchers are concerned about how food web properties, such as diversity, may stabilize food webs. Several properties have been proposed to stabilize food webs, the most prominent of which are lack of omnivory, greater compartmentalization/modularity and coherence, and greater generality in natural enemies coupled with greater vulnerability of their prey (SI Glossary). In addition, the growing body of research on general interaction networks, of which food webs are an example, has opened questions about the connectedness and clustering of populations and substructuring among a small number of populations. These will be discussed in turn in the following.
Early theoretical investigations suggested that omnivory would destabilize food webs, and therefore, omnivory should be rare (Pimm 1979). Omnivory is the feeding on multiple trophic levels and is estimated with an omnivory index (OI), which is equal to the variance of the trophic levels of a consumer’s food groups. The OI is zero when all feeding occurs from the same trophic level and increases with the variety of trophic levels consumed. Contrary to the theoretical prediction, empirical work has shown that omnivory is common in food webs (e.g., Polis 1991). In studies of arthropod natural enemies, intraguild predation, which is a form of omnivory, has been commonly found (Vance-Chalcraft et al. 2007) and has also been common in natural enemy sink webs based on molecular gut content analysis (e.g., Paula et al. 2016, 2022a). More recent theoretical research, stimulated by the contradiction, has found multiple conditions under which omnivory is stabilizing, especially in complex food webs (Kratina et al. 2012).
Food web compartmentalization/modularity is suggested to confer stability. Compartmentalization is the organization of the populations into groups that interact more commonly within groups than between groups. A fully compartmentalized community has small groups of populations interacting with each other with no interactions between populations in different compartments. A community with no compartmentalization has random interactions among populations. Pimm (1979) suggested that an intermediate level of compartmentalization may be most stable. This may be because at intermediate levels, cross-compartment interactions reduce the variation within compartments (McCann et al. 1998; Rooney et al. 2006). Recently, formal methods for detecting compartmentalization in food webs have been developed (Guimarà et al. 2010). These methods have revealed significant compartmentalization in empirical food webs (Fig. 2), indicating that empirical food webs are organized into intermediate levels of compartmentalization, which may enhance stability (McCann et al. 1998; Rooney et al. 2006).

Adjacency matrix showing compartmentalization in an empirical food web. Prey compartments (different colors among rows) and predator compartments (different colors among columns) correspond to groups of species with similar predators (for prey) or prey (for predators). Black squares indicate the existence of a predator–prey relation and white squares its absence. The adjacency matrix was rearranged to reveal the block structure of the compartments. Redrawn from Guimerà et al. (2010)
Food web coherence is the variation in the trophic levels of the different prey consumed by a natural enemy and is measured by the standard deviation of these values (q, Johnson et al. 2014). In a perfectly coherent food web, all consumption occurs on prey exactly one trophic level removed from the predator. In other words, all herbivores feed only on plants, and all predators feed either only on herbivores or only on other predators, and there is no omnivory, cannibalism, or intraguild predation. In this case, q is 0. All perfectly coherent food webs are stable (Johnson et al. 2014). As maximum coherence occurs for q = 0, q is a measure of food web incoherence. Although several simple food webs are nearly coherent (Johnson et al. 2014), some simple sink webs constructed from molecular gut content analysis have larger q (Paula et al., unpublished), suggesting that in some cases these sink food webs may not be stable.
Vulnerability and generality are food web properties that have been examined for some time (Goldwasser and Roughgarden 1993). Vulnerability is related to the number of natural enemies for a given prey, and food webs with high vulnerability mean that prey are susceptible to predation from many predator populations. Generality is related to the number of prey populations for a given natural enemy, which is the ecological prey range and is useful for assessing the potential adverse effects of a predator. Gross et al. (2008) showed that higher generality in the top natural enemies coupled with higher vulnerability in the intermediate trophic levels, such as herbivores and the prey of the top natural enemies leads to greater food web stability. Molecular sink webs may provide unbiased estimates of generality but may underestimate vulnerability, because some of the predators of the prey may be missing from the samples. In a molecular sink food web, vulnerability varied with prey population and time of the season, while generality varied with predator population and slight variation over time of season (Fig. 3). These results indicate that most of the prey populations have high vulnerability to the predator populations and most of the predator populations have high generality for the prey populations, suggesting that the food web may be stable.

A Vulnerability of six populations of extraguild prey to four predator populations and b generality of four populations of predators on six species of prey from May to September (Paula, unpublished data). A.cit, Aphis citricidus; A.sol, Aphis solanella; B.bra, Brevicoryne brassicae; L.pse, Lipaphis pseudobrassicae; M.per, Myzus persicae; U.amb, Uroleucon ambrosiae; C.san, Cycloneda sanguinea; E.con, Eriopis connexa; H.axy, Harmonia axyridis; Hi.con, Hippodamia convergens
Many general interaction networks have “small world” topologies. In a small world network, nodes (populations) are more closely connected to other nodes than expected at random, and nodes tend to interact primarily with a small cluster of nodes. In a small world topology, interactions tend to be clustered or compartmentalized, but there are enough connections among clusters that it takes only a few connections for clusters to influence each other. For example, Milgram (1967) suggested that every person in the world is only “six degrees of separation” from every other human, which means, for example, a disease can transmit throughout the world via transmission to only six people. Watts and Strogatz (1998) formalized tests to determine if a network exhibits “small world” topologies. The first is evaluated by the characteristic path length (e.g., six people), which in a food web is the average of the smallest number of linkages connecting a prey to a predator (SI Glossary). The second is evaluated by the clustering coefficient (SI Glossary), which in a sink food web is the fraction of intraguild predation linkages among the potential intraguild linkages for the predators feeding on the same prey. A short characteristic path length may result in a rapid spread of a perturbation (e.g., species invasion or extirpation) through the food web (Watts and Strogatz 1998; Williams et al. 2002) but a high clustering coefficient (e.g., close to 1) may be associated with high connectance (SI Glossary) and greater resilience within clusters to perturbation (Dunne et al. 2002). Molecular sink food webs have short critical path lengths but do not exhibit high clustering (Paula et al., unpublished), although few have been examined to know if these results are general.
A recently identified network property is a network motif (SI Glossary). A network motif is a substructure of the network and is a recurring pattern of interconnections among a small number of nodes that occurs more commonly than random (Fig. 4, Milo et al. 2002). There are several possible motifs in food webs involving three populations. The tri-trophic interaction was more common than random in nearly all empirical food webs, which corresponds to the naive view that communities are organized into food chains (Stouffer et al. 2007). Food webs divided into two motif types. The larger group of food webs, which includes most of the terrestrial food webs, had an overrepresentation of omnivory/intraguild predation and an underrepresentation of shared predation and apparent competition (Fig. 4). The smaller group of food webs had low omnivory/intraguild predation and high shared predation and apparent competition. The reason for the difference in these two groups of food webs is not known. As intraguild predation has been commonly found in molecular sink food webs (Paula et al. 2016; Andow and Paula, unpublished), elaboration of molecular sink webs will probably increase the number of the first group of food webs.

Common three-species motifs in empirical food webs, showing the motif in black and white and all examples of the motif embedded in a food web with red linkages. A motif must contain all indicated linkages and only those indicated linkages. a Tri-trophic interaction; b omnivory/intraguild predation; c shared predation; d apparent competition
Future Avenues
Biological control research can benefit tremendously of the advances in DNA-based methods, such as HTS, not only for detection of species and their interactions, but also to assess the strength of such interactions. The capacity of detecting multiple and previously unknown interactions provided by metabarcoding and Lazaro, is enabling an unpreceded ability to construct complex multitrophic food webs at various spatial and temporal resolutions, which otherwise would be impractical to obtain from natural systems. These have direct implications for the understanding of community dynamics and improvement of predictions of the biological control potential to regulate pests and not have adverse impacts on non-targets. However, there remains considerable room for fundamental research to control for false-positive and -negative identifications and to produce relative read abundances that provide accurate quantitative estimates of predation rates.
References
Agustí N, De Vicente MC, Gabarra R (1999) Development of sequence amplified characterized region (SCAR) markers of Helicoverpa armigera: a new polymerase chain reaction-based technique for predator gut analysis. Mol Ecol 8(9):1467–1474
Agustí N, Symondson WOC (2001) Molecular diagnosis of predation. Antenna 25:250–253
Aird D, Ross MG, Chen WS et al (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12:R18
Alberdi A, Aizpurua O, Gilbert MT, Bohmann K (2018) Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods Ecol Evol 9(1):134–147
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Amend AS, Seifert KA, Bruns TD (2010) Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Mol Ecol 19:5555–5565
Barazzoni R, Short KR, Nair KS (2000) Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem 275(5):3343–3347
Barlow JT, Bogatyrev SR, Ismagilov RF (2020) A quantitative sequencing framework for absolute abundance measurements of mucosal and lumenal microbial communities. Nat Commun 11(1):1–3
Berlow EL, Neutel AM, Cohen JE, De Ruiter PC, Ebenman BO, Emmerson M, Fox JW, Jansen VA, Iwan Jones J, Kokkoris GD, Logofet D (2004) Interaction strengths in food webs: issues and opportunities. J an Ecol 73(3):585–598
Berry D, Mahfoudh KB, Wagner M, Loy A (2011) Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol 77:7846–7849
Birkhofer K, Bylund H, Dalin P, Ferlian O, Gagic V, Hambäck PA, ... Jonsson M (2017). Methods to identify the prey of invertebrate predators in terrestrial field studies. Ecol Evol 7(6):1942-1953
Bista I, Carvalho GR, Tang M et al (2018) Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples. Mol Ecol Resour 18:1020–1034
Boreham PFL, Ohiagu CE (1978) The use of serology in evaluating invertebrate predator-prey relationships: a review. Bull Entomol Res 68:171–194
Boyer F, Mercier C, Bonin A, Le Bras Y, Taberlet P, Coissac E (2016) Obitools: a unix-inspired software package for DNA metabarcoding. Mol Ecol Resour 16(1):176–182
Braukmann TW, Ivanova NV, Prosser SW, Elbrecht V, Steinke D, Ratnasingham S, ... & Hebert PD (2019) Metabarcoding a diverse arthropod mock community. Mol Ecol Resour 19(3):711-727
Breitwieser FP, Baker DN, Salzberg SL (2018) KrakenUniq: confident and fast metagenomics classification using unique k-mer counts. Genome Biol 19:1–10
Bridge PD, Roberts PJ, Spooner BM, Panchal G (2003) On the unreliability of published DNA sequences. New Phytol 160(1):43–48
Brook MM, Proske HO (1946) Precipitin test for determining natural insect predators of immature mosquitoes. J Natl Malar Soc 5:45–56
Brown DS, Jarman SN, Symondson WO (2012) Pyrosequencing of prey DNA in reptile faeces: analysis of earthworm consumption by slow worms. Mol Ecol Resour 12(2:259–266.
Calder C, Harwood JD, Symondson WO (2005) Detection of scavenged material in the guts of predators using monoclonal antibodies: a significant source of error in measurement of predation? Bull Entomol Res 95:57–62
Caporaso JG, Kuczynski J, Stombaugh J et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Carlsen T, Aas AB, Lindner D, Vrålstad T, Schumacher T, Kauserud H (2012) Don’t make a mistake: is tag switching an overlooked source of error in amplicon pyrosequencing studies? Fungal Ecol 5(6):747–749
Champlot S, Berthelot C, Pruvost M, Bennett EA, Grange T, Geigl EM (2010) An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS ONE 5(9):e13042
Chen Y, Giles KL, Payton ME, Greenstone MH (2000) Identifying key cereal aphid predators by molecular gut analysis. Mol Ecol 9:1887–1898
Clare EL (2014) Molecular detection of trophic interactions: emerging trends, distinct advantages, significant considerations and conservation applications. Evol Appl 7(9):1144–1157
Clarke LJ, Soubrier J, Weyrich LS, Cooper A (2014) Environmental metabarcodes for insects: in silico PCR reveals potential for taxonomic bias. Mol Ecol Resour 14:1160–1170
Cline J, Braman JC, Hogrefe HH (1996) PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res 24:3546–3551
Cohen JE (1978) Food Webs and Niche Space. Princeton University Press
Cotterill M, Harris SE, Collado Fernandez E, Lu J, Huntriss JD, Campbell BK, Picton HM (2013) The activity and copy number of mitochondrial DNA in ovine oocytes throughout oogenesis in vivo and during oocyte maturation in vitro. Mol Hum Reprod 19(7):444–450
Crampton-Platt A, Timmermans MJ, Gimmel ML, Kutty SN, Cockerill TD, Vun Khen C, Vogler AP (2015) Soup to tree: the phylogeny of beetles inferred by mitochondrial metagenomics of a Bornean rainforest sample. Mol Biol Evol 32(9):2302–2316
Davey JS, Vaughan IP, King RA, Bell JR, Bohan DA, Bruford MW, Holland JM, Symondson WOC (2013) Intraguild predation in winter wheat: prey choice by a common epigeal carabid consuming spiders. J Appl Ecol 50(1):271–279
De Barba M, Miquel C, Boyer F, Mercier C, Rioux D, Coissac E, Taberlet P (2014) DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: application to omnivorous diet. Mol Ecol Resour 14:306–323
Deagle B, Eveson J, Jarman S (2006) Quantification of damage in DNA recovered from highly degraded samples - A case study on DNA in faeces. Front Zool 3:11
Deagle BE, Tollit DJ (2007) Quantitative analysis of prey DNA in pinniped faeces: potential to estimate diet composition? Conserv Genet 8:743–747
Deagle BE, Kirkwood R, Jarman SN (2009) Analysis of Australian fur seal diet by pyrosequencing prey DNA in faeces. Mol Ecol 18(9):2022–2038
Deagle BE, Thomas AC, Shaffer AK, Trites AW, Jarman SN (2013) Quantifying sequence proportions in a DNA-based diet study using Ion Torrent amplicon sequencing: which counts count? Mol Ecol Resour 13:620–633
Deagle BE, Jarman SN, Coissac E, Pompanon F, Taberlet P (2014) DNA metabarcoding and the COI marker: not a perfect match. Biol Lett 10:20140562
Deagle BE, Thomas AC, McInnes JC, Clarke LJ, Vesterinen EJ et al (2019) Counting with DNA in metabarcoding studies: how should we convert sequence reads to dietary data? Mol Ecol 28:391–440
Dempster JP (1960) A quantitative study of the predators on the eggs and larvae of the broom beetle, Phytodecta olivacea Forster, using the precipitin test. J an Ecol 29:149–167
Dennison DF, Hodkinson ID (1983) Structure of the predatory beetle community in a woodland soil ecosystem. I Prey Selection Pedobiologia 25:109–115
Di Muri C, Lawson Handley L, Bean CW, Li J, Peirson G, Sellers GS, Walsh K, Watson HV, Winfield IJ, Hänfling B (2020) Read counts from environmental DNA (eDNA) metabarcoding reflect fish abundance and biomass in drained ponds. Metabarcoding Metagenom 4:97–112
Dopheide A, Xie D, Buckley TR, Drummond AJ, Newcomb RD (2019) Impacts of DNA extraction and PCR on DNA metabarcoding estimates of soil biodiversity. Meth Ecol Evol 10(1):120–133
Dupuis JR, Roe AD, Sperling FA (2012) Multi-locus species delimitation in closely related animals and fungi: one marker is not enough. Mol Ecol 21(18):4422–4436
Elbrecht V, Leese F (2015) Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass - sequence relationships with an innovative metabarcoding protocol. PLoS ONE 10(7):e0130324
Elbrecht V, Taberlet P, Dejean T et al (2016) Testing the potential of a ribosomal 16S marker for DNA metabarcoding of insects. PeerJ 4:e1966
Elbrecht V, Leese F (2017) Validation and development of COI metabarcoding primers for freshwater macroinvertebrate bioassessment. Front Environ Sci 5:11
Engelbrektson A, Kunin V, Wrighton K et al (2010) Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J 4:642–647
Ferlian O, Scheu S, Pollierer MM (2012) Trophic interactions in centipedes (Chilopoda, Myriapoda) as indicated by fatty acid patterns: variations with life stage, forest age and season. Soil Biol Biochem 52:33–42
Ficetola GF, Miaud C, Pompanon F, Taberlet P (2008) Species detection using environmental DNA from water samples. Biol Lett 4:423–425
Ficetola GF, Coissac E, Zundel S et al (2010) An in silico approach for the evaluation of DNA barcodes. BMC Genomics 11:434
Ficetola GF, Pansu J, Bonin A, Coissac E, Giguet-Covex C, De Barba M, ... Taberlet, P (2015) Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol Ecol Resour 15(3):543-556
Ficetola GF, Taberlet P, Coissac E (2016) How to limit false positives in environmental DNA and metabarcoding? Mol Ecol Resour 16:604–607
Fichter BL, Stephen WP (1981) Time related decay in prey antigens ingested by the predator Podisus maculiventris (Hemiptera, Pentatomidae) as detected by ELISA. Oecologia 51:404–407
Fichter BL, Stephen WP (1984) Time-related decay of prey antigens ingested by arboreal spiders as detected by ELISA. Environ Entomol 13(6):1583–1587
Floyd R, Abebe E, Papert A, Blaxter M (2002) Molecular barcodes for soil nematode identification. Mol Ecol 11:839–850
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3(5):294–299
Foltan P, Sheppard S, Konvicka M, Symondson WOC (2005) The significance of facultative scavenging in generalist predator nutrition: detecting decayed prey in the guts of predators using PCR. Mol Ecol 14:4147–4158
Forsythe TG (1982) Feeding mechanisms of certain ground beetles (Coleoptera: Carabidae). The Coleopterists’ Bulletin, 26–73
Fournier V, Hagler J, Daane K et al (2008) Identifying the predator complex of Homalodisca vitripennis (Hemiptera: Cicadellidae): a comparative study of the efficacy of an ELISA and PCR gut content assay. Oecologia 157:629–640
Fox CJS, MacLellan CR (1956) Some Carabidae and Staphylinidae shown to feed on a wireworm, Agriotes sputator (L), by the precipitin test. Canad Ent 88:228–231
Frank SD, Wratten SD, Sandhu HS, Shrewsbury PM (2007) Video analysis to determine how habitat strata affects predator diversity and predation of Epiphyas postvittana (Lepidoptera: Tortricidae) in a vineyard. Biol Control 41(2):230–236
Furlong MJ (2015) Knowing your enemies: Integrating molecular and ecological methods to assess the impact of arthropod predators on crop pests. Insect Sci 22(1):6–19
Gagnon A-È, Heimpel GE, Brodeur J (2011a) The ubiquity of intraguild predation among predatory arthropods. PLoS ONE 6(11):e28061
Gagnon AÈ, Doyon J, Heimpel GE, Brodeur J (2011b) Prey DNA detection success following digestion by intraguild predators: influence of prey and predator species. Mol Ecol Resour 11(6):1022–1032
Geiger F, Bengtsson J, Berendse F, Weisser et al (2010) Persistent negative effects of pesticides on biodiversity and biological control potential on European farmland. Basic Appl Ecol 11(2):97–105
Gibson J, Shokralla S, Porter TM, King I, van Konynenburg S, Janzen DH et al (2014) Simultaneous assessment of the macrobiome and microbiome in a bulk sample of tropical arthropods through DNA metasystematics. PNAS 111:8007–8012
Giguet-Covex C, Pansu J, Arnaud F et al (2014) Long live-stock farming history and human landscape shaping revealed by lake sediment DNA. Nat Commun 5:3211
Goldwasser L, Roughgarden J (1993) Construction of a large Caribbean food web. Ecology 74:1216–1233
Gómez-Rodríguez C, Crampton-Platt A, Timmermans MJ, Baselga A, Vogler AP (2015) Validating the power of mitochondrial metagenomics for community ecology and phylogenetics of complex assemblages. Methods Ecol Evol 6:883–894
Greenstone MH (1977) A passive haemagglutination inhibition assay for the identification of stomach contents of invertebrate predators. J Appl Ecol 14:457–464
Greenstone MH, Hunt JH (1993) Determination of prey antigen half-life in Polistes metricus using a monoclonal antibody-based immunodot assay. Entomol Exp Appl 68(1):1–7
Greenstone MH (1996) Serological analysis of arthropod predation: past, present and future. In: Symondson WOC, Liddell E (eds) The ecology of agricultural pests: biochemical approaches. Chapman and Hall, London, UK, pp 265–300
Greenstone MH, Rowley DL, Weber DC, Payton ME, Hawthorne DJ (2007) Feeding mode and prey detectability half-lives in molecular gut-content analysis: an example with two predators of the Colorado potato beetle. Bull Entomol Res 97(2):201–209
Greenstone MH, Szendrei Z, Payton ME, Rowley DL, Coudron TC, Weber DC (2010) Choosing natural enemies for conservation biological control: use of the prey detectability half-life to rank key predators of Colorado potato beetle. Entomol Exp Appl 136(1):97–107
Greenstone MH, Weber DC, Coudron TA, Payton ME, Hu JS (2012) Removing external DNA contamination from arthropod predators destined for molecular gut-content analysis. Mol Ecol Resour 12(3):464–469
Greenstone MH, Payton ME, Weber DC, Simmons AS (2013) The detectability half-life in arthropod predator–prey research: what it is, why we need it, how to measure it, and how to use it. Mol Ecol 23:3799–3813
Guimerà R, Stouffer DB, Sales-Pardo M, Leicht EA, Newman MEJ, Amaral LA (2010) Origin of compartmentalization in food webs. Ecology 91(10):2941–2951
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G et al (2011) Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21(3):494–504
Hagler JR (2006) Development of an immunological technique for identifying multiple predator–prey interactions in a complex arthropod assemblage. Ann Appl Biol 149(2):153–165
Hagler JR (2019) It’s gut check time! A universal food immunomarking technique for studying arthropod feeding activities. Ann Entomol Soc Am 112(3):211–219
Hagler JR, Naranjo SE (1994) Determining the frequency of heteropteran predation on sweetpotato whitefly and pink bollworm using multiple ELISAs. Entomol Exp Appl 72(1):59–66
Hagler JR, Naranjo SE (1997) Measuring the sensitivity of an indirect predator gut content ELISA: detectability of prey remains in relation to predator species, temperature, time, and meal size. Biol Control 9(2):112–119
Hagler JR, Blackmer F (2013) Identifying inter-and intra-guild feeding activity of an arthropod predator assemblage. Ecol Entomol 38(3):258–271
Hall RR, Downe AER, MacLellan CR, West AS (1953) Evaluation of insect predator-prey relationships by precipitin test studies. Mosq News 13:199–204
Hance T, Rossignol R (1983) Essai de quantification de la prédation des Carabidae par le test ELISA. Medelingen Van De Faculteit Landbouwwetenschappen Rijksuniversiteit Gent 48:475
Harper GL, King RA, Dodd CS, Harwood JD, Glen DM, Bruford MW, Symondson WOC (2005) Rapid screening of invertebrate predators for multiple prey DNA targets. Mol Ecol 14:819–827
Harper GL, Sheppard SK, Harwood JD et al (2006) Evaluation of temperature gradient gel electrophoresis for the analysis of prey DNA within the guts of invertebrate predators. Bull Entomol Res 96:295–304
Harris JK, Sahl JW, Castoe TA et al (2010) Comparison of normalization methods for construction of large, multiplex amplicon pools for next-generation sequencing. Appl Environ Microbiol 76:3863–3868
Harwood JD, Phillips SW, Sunderland KD, Symondson WOC (2001) Secondary predation: quantification of food chain errors in an aphid–spider–carabid system using monoclonal antibodies. Mol Ecol 10(8):2049–2057
Harwood JD, Obrycki JJ (2005) Web-construction behavior of linyphiid spiders (Araneae, Linyphiidae): competition and co-existence within a generalist predator guild. J Ins Behav 18(5):593–607
Hassell MP, May RM (1973) Stability in insect host–parasite models. J Anim Ecol 42:693–726. https://doi.org/10.2307/3133
Hebert PDN, Cywinska A, Ball SL, deWaard JR (2003a) Biological identifications through DNA barcodes. Proc Biol Sciences 270:313–321
Hebert PDN, Ratnasingham S, deWaard JR (2003b) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci 270:96–99
Hsieh TC, Ma KH, Chao A (2016) iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Method Ecol Evol 7:1451–1456
Holling CS (1959) Some characteristics of simple types of predation and parasitism. Can Entomol 91:385–398
Holmes PR (1984) A field study of the predators of the grain aphid, Sitobion ailenae (F) (Hemiptera: Aphididae), in winter wheat. Bull Entomol Res 74:623–631
Hoogendoorn M, Heimpel GE (2001) PCR-based gut content analysis of insect predators: using ribosomal ITS-1 fragments from prey to estimate predation frequency. Mol Ecol 10:2059–2067
Hosseini R, Schmidt O, Keller MA (2008) Factors affecting detectability of prey DNA in the gut contents of invertebrate predators: a polymerase chain reaction-based method. Entomol Exp Appl 126:194–202
Huson DH, Mitra S, Ruscheweyh H-J et al (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21:1552–1560
Ihrmark K, Bödeker IT, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandström-Durling M, Clemmensen KE, Lindahl BD (2012) New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82(3):666–677
Ingerson-Mahar J (2002) Relating diet and morphology in adult carabid beetles. In: The Agroecology of Carabid Beetles (ed. Holland J), pp. 111–136. Intercept, Andover, UK
Jaric M, Segal J, Silva-Herzog E et al (2013) Better primer design for metagenomics applications by increasing taxonomic distinguishability. BMC Proc 7:S4
Jarman SN, Redd KS, Gales NJ (2005) Group-specific primers for amplifying DNA sequences that identify Amphipoda, Cephalopoda, Echinodermata, Gastropoda, Isopoda, Ostracoda and Thoracica. Mol Ecol 6:268–271
Ji Y, Ashton L, Pedley SM, Edwards DP, Tang Y, Nakamura A et al (2013) Reliable, verifiable and efficient monitoring of biodiversity via metabarcoding. Ecol Lett 16:1245–1257
Ji Y, Huotari T, Roslin T, Schmidt NM, Wang J, Yu DW, Ovaskainen O (2020) SPIKEPIPE: a metagenomic pipeline for the accurate quantification of eukaryotic species occurrences and intraspecific abundance change using DNA barcodes or mitogenomes. Mol Ecol Resour 20(1):256–267
Johnson S, Domínguez-García V, Donetti L, Muñoz MA (2014) Trophic coherence determines food-web stability. PNAS 111(50):17923–17928
Jones MG (1979) Abundance of aphids on cereals from before 1973 to 1977. J Appl Ecol 16:1–22
Juen A, Traugott M (2005) Detecting predation and scavenging by DNA gut-content analysis: a case study using a soil insect predator-prey system. Oecologia 142:344–352
Juen A, Hogendoorn K, Ma G, Schmidt O, Keller MA (2012) Analyzing the diets of invertebrate predators using terminal restriction fragments. J Pest Sci 85(1):89–100
Kaestner A (1993) Araneomorphae. In: Lehrbuch der Speziellen Zoologie, 4th edn. (eds HE Gruner, M Moritz, W Dunger), 244– 263. Spektrum Akademischer Verlag Fischer, Jena, 1279 pp
Kamenova S, Mayer R, Rubbmark OR, Coissac E, Plantegenest M, Traugott M (2018) Comparing three types of dietary samples for prey DNA decay in an insect generalist predator. Mol Ecol Resour 18(5):966–973
Kartzinel TR, Chen PA, Coverdale TC, Erickson DL, Kress WJ, Kuzmina ML et al (2015) DNA metabarcoding illuminates dietary niche partitioning by African large herbivores. PNAS 112(26):8019–8024
Kebschull JM, Zador AM (2015) Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res 43(21):e143
King R, Read D, Traugott M, Symondson W (2008) Molecular analysis of predation: a review of best practice for DNA-based approaches. Mol Ecol 17:947–963
King RA, Moreno-Ripoll R, Agustí N, Shayler SP, Bell JR, Bohan DA, Symondson WO (2011) Multiplex reactions for the molecular detection of predation on pest and nonpest invertebrates in agroecosystems. Mol Ecol Resour 11(2):370–373
Kobayashi N, Tamura K, Aotsuka T (1999) PCR error and molecular population genetics. Biochem Genet 37:317–321
Kowalczyk R, Taberlet P, Coissac E, Valentini A, Miquel C, Kamiński T, Wójcik JM (2011) Influence of management practices on large herbivore diet-case of European bison in Białowieża Primeval Forest (Poland). Forest Ecol Manag 261(4):821–828
Kratina P, LeCraw RM, Ingram T, Anholt BR (2012) Stability and persistence of food webs with omnivory: is there a general pattern? Ecosphere 3:1–18
Krehenwinkel H, Kennedy S, Pekár S, Gillespie RG (2017) A cost-efficient and simple protocol to enrich prey DNA from extractions of predatory arthropods for large-scale gut content analysis by Illumina sequencing. Methods Ecol Evol 8(1):126–134
Kwok S, Kellogg DE, McKinney N, Spasic D, Goda L, Levenson C, Sninsky JJ (1990) Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res 18(4):999–1005
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P (2010) Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123
Kuperstein ML (1979) Estimating carabid effectiveness in reducing the Sunn pest, Eurygaster integriceps Puton (Heteroptera: Scutelleridae) in the U.S.S.R. Ent Soc Am Misc Publication 11:80–84
Kuusk AK, Ekbom B (2010) Lycosid spiders and alternative food: feeding behavior and implications for biological control. Biol Control 55(1):20–26
Lahoz-Monfort JJ, Guillera-Arroita G, Tingley R (2016) Statistical approaches to account for false-positive errors in environmental DNA samples. Mol Ecol Resour 16:673–685
Lamb PD, Hunter E, Pinnegar JK, Creer S, Davies RG, Taylor MI (2018) How quantitative is metabarcoding: a meta-analytical approach. Mol Ecol 28(2):420–430
Layman CA, Arrington DA, Montana CG, Post DM (2007) Can stable isotope ratios provide for community-wide measures of trophic structure? Ecology 88:42–48
Lefort MC, Wratten S, Cusumano A, Varennes YD, Boyer S (2017) Disentangling higher trophic level interactions in the cabbage aphid food web using high-throughput DNA sequencing. Metabarcoding Metagenom 1:13709
Leite LAR (2012) Mitochondrial pseudogenes in insect DNA barcoding: differing points of view on the same issue. Biota Neotrop 12(3):301–308
Leray M, Yang JY, Meyer CP et al (2013) A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front Zool 10:34
Levins R (1970) Complex systems. In: Waddington CH (ed) Towards a theoretical biology: 3 Drafts. Edinburgh Univ. Press, Edinburgh, pp 73–88
Li K, Tian J, Wang QX, Chen Q, Chen M, Wang H, Zhou YX, Peng YF, Xiao JH, Ye GY (2011) Application of a novel method PCR-ligase detection reaction for tracking predator–prey trophic links in insect-resistant GM rice ecosystem. Ecotoxicology 20:2090–2100
Li X, Yang Y, Henry RJ, Rossetto M, Wang Y, Chen S (2015) Plant DNA barcoding: from gene to genome. Biol Rev 90(1):157–166
Linard B, Crampton-Platt A, Moriniere J, Timmermans MJ, Andujar C, Arribas P, Vogler AP (2018) The contribution of mitochondrial metagenomics to large-scale data mining and phylogenetic analysis of Coleoptera. Mol Phylogenet Evol 128:1–11
Lister A, Usher MB, Block W (1987) Description and quantification of field attack rates by predatory mites: an example using an electrophoresis method with a species of Antarctic mite. Oecologia 72:185–191
Liu M, Clarke LJ, Baker SC, Jordan GJ, Burridge CP (2020) A practical guide to DNA metabarcoding for entomological ecologists. Ecological Entomology 45(3):373–385
Lövei GL 1986. The use of biochemical methods in the study of carabid feeding: the potential of isoenzyme analysis and ELISA. In: Den Boer, P.J, Grüm,L, Szyszko, J.,(Eds.) Feeding behaviour and accessibility of food for carabid beetles. Proceedings of the 5th Meeting of European Carabidologists. Agricultural University Press, Warsaw, pp. 21–27.
Lövei GL, Monostori É, Andó I (1985) Digestion rate in relation to starvation in the larva of a carabid predator. Poecilus Cupreus Entomologia Experimentalis Et Applicata 37(2):123–127
Lundgren JG, Ellsbury ME, Prischmann DA (2009) Analysis of the predator community of a subterranean herbivorous insect based on polymerase chain reaction. Ecol Appl 19:2157–2166
Lundgren JG, Fergen JK (2011) Enhancing predation of a subterranean insect pest: a conservation benefit of winter vegetation in agroecosystems. Appl Soil Ecol 51:9–16
Martin DL, Ross RM, Quetin LB, Murray AE (2006) Molecular approach (PCR-DGGE) to diet analysis in young Antarctic krill Euphausia superba. Mar Ecol Prog Ser 319:155–165
May RM (1972) Will a large complex system be stable? Nature 238(5364):413–414
McCann K, Hastings A, Huxel GR (1998) Weak trophic interactions and the balance of nature. Nature 395:794–798
Memmott J, Martinez ND, Cohen JE (2000) Predators, parasitoids and pathogens: species richness, trophic generality and body sizes in a natural food web. J Anim Ecol 69(1):1–15
Milgram S (1967) The Small World Problem Psychology Today 1:61–67
Miller MC (1981) Evaluation of enzyme-linked immunosorbent assay of narrow- and broad-spectrum anti-adult southern pine beetle serum. Ann Entomol Soc Am 74:279–282
Miller DA, Nichols JD, McClintock BT, Grant EHC, Bailey LL et al (2011) Improving occupancy estimation when two types of observational error occur: non-detection and species misidentification. Ecology 92:1422–1428
Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U (2002) Network motifs: simple building blocks of complex networks. Science 298(5594):824–827
Muller CB, Adriaanse ICT, Belshaw R, Godfray HCJ (1999) The structure of an aphid–parasitoid community. J Anim Ecol 68(2):346–370
Mullis KB (1990) The unusual origin of the polymerase chain reaction. Sci Am 262(4):56–65
Munch K, Boomsma W, Willerslev E et al (2008a) Fast phylogenetic DNA barcoding. Philos Trans R Soc B 363:3997–4002
Munch K, Boomsma W, Huelsenbeck JP, Willerslev E, Nielsen R (2008b) Statistical assignment of DNA sequences using Bayesian phylogenetics. Syst Biol 57(5):750–757
Murray RA, Solomon MG (1978) A rapid technique for analysing diets of invertebrate predators by electrophoresis. Annals of Applied Biology 90(1):7–10
Murray DC, Bunce M, Cannell BL et al (2011) DNA-based faecal dietary analysis: a comparison of qPCR and high throughput sequencing approaches. PLoS ONE 6:e25776
Murray DC, Coghlan ML, Bunce M (2015) From benchtop to desktop: important considerations when designing amplicon sequencing workflows. PLoS ONE 10(4):e0124671
Nakamura M, Nakamura K (1977) Population dynamics of the chestnut gall wasp, Dryocosmus kuriphilus Yasumatsu (Hymenoptera: Cynipidae). Oecologia 27(2):97–116
Naranjo SE, Hagler JR (2001) Toward the quantification of predation with predator gut immunoassays: a new approach integrating functional response behavior. Biol Control 20(2):175–189
Neby M, Kamenova S, Devineau O, Ims RA, Soininen EM (2021) Issues of under-representation in quantitative DNA metabarcoding weaken the inference about diet of the tundra vole Microtus oeconomus. PeerJ 9:e11936
Nichols RV, Vollmers C, Newsom LA, Wang Y, Heintzman PD, Leighton M, Green RE, Shapiro B (2018) Minimizing polymerase biases in metabarcoding. Mol Ecol Resour 18(5):927–939
Nguyen NH, Smith D, Peay K, Kennedy P (2015) Parsing ecological signal from noise in next generation amplicon sequencing. New Phytol 205(4):1389–1393
O’Neil RJ, Stimac JL (1988) Model of arthropod predation on velvetbean caterpillar (Lepidoptera: Noctuidae) larvae in soybeans. Environ Entomol 17:983–987
Pääbo S, Irwin DM, Wilson AC (1990) DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem 265(8):4718–4721
Paula DP, Linard B, Andow DA, Sujii ER, Pires CS, Vogler AP (2015) Detection and decay rates of prey and prey symbionts in the gut of a predator through metagenomics. Mol Ecol Resour 15(4):880–892
Paula DP, Linard B, Crampton-Platt A, Srivathsan A, Timmermans MJ, Sujii ER, Pires CS, Souza LM, Andow DA, Vogler AP (2016) Uncovering trophic interactions in arthropod predators through DNA shotgun-sequencing of gut contents. PLoS ONE 11(9):e0161841
Paula DP, Barros SKA, Pitta RM, Barreto MR, Togawa RC, Andow DA (2022a) Metabarcoding versus mapping unassembled shotgun reads for identification of prey consumed by arthropod epigeal predators. GigaScience 11:1–13
Paula DP, Timbó RV, Togawa RC, Vogler AP, Andow DA (2022b) Quantitative prey species detection in predator guts across multiple trophic levels by mapping unassembled shotgun reads. Mol Ecol Resour. https://doi.org/10.1111/1755-0998.13690
Pendleton RC, Grundmann AW (1954) Use of P32 in tracing some insect-plant relationships of the thistle Cirsium undulatum. Ecology 35:187–191
Peterson JA, Burkness EC, Harwood JD, Hutchison WD (2018) Molecular gut-content analysis reveals high frequency of Helicoverpa zea (Lepidoptera: Noctuidae) consumption by Orius insidiosus (Hemiptera: Anthocoridae) in sweet corn. Bio Control 121:1–7
Pimm SL (1979) The structure of food webs. Theor Popul Biol 16:144–158
Piñol J, San Andrés V, Clare EL, Mir G, Symondson WOC (2014a) A pragmatic approach to the analysis of diets of generalist predators: the use of next-generation sequencing with no blocking probes. Mol Ecol Resour 14(1):18–26
Piñol J, Mir G, Gomez-Polo P, Agustí N (2015) Universal and blocking primer mismatches limit the use of high throughput DNA sequencing for the quantitative metabarcoding of arthropods. Mol Ecol Resour 15:1–12
Piñol J, Senar MA, Symondson WO (2018) The choice of universal primers and the characteristics of the species mixture determines when DNA metabarcoding can be quantitative. Mol Ecol 28:407–419
Piñol J, San Andres V, Clare EL, Mir G, Symondson WOC (2014b) A pragmatic approach to the analysis of diets of generalist predators: the use of next-generation sequencing with no blocking probes. Mol Ecol Resour 14:18–26
Pinto AJ, Raskin L (2012) PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS ONE 7:e43093
Polis GA (1991) Complex desert food webs: an empirical critique of food web theory. Amer Natur 138:123–155
Pommier T, Neal PR, Gasol JM et al (2010) Spatial patterns of bacterial richness and evenness in the NW Mediterranean Sea explored by pyrosequencing of the 16S rRNA. Aquat Microb Ecol 61:212–224
Pompanon F, Deagle BE, Symondson WOC, Brown DS, Jarman SN, Taberlet P (2012) Who is eating what: diet assessment using next generation sequencing. Mol Ecol 21:1931–1950
Porter TM, Gibson JF, Shokralla S, Baird DJ, Golding GB, Hajibabaei M (2014) Rapid and accurate taxonomic classification of insect (class Insecta) cytochrome c oxidase subunit 1 (COI) DNA barcode sequences using a naïve Bayesian classifier. Mol Ecol Resour 14(5):929–942
Post DM (2002) Using stable isotopes to estimate trophic position: models, methods, and assumptions. Ecology 83:703–718
Porazinska DL, Sung W, Giblin-Davis RM, Thomas WK (2010) Reproducibility of read numbers in high-throughput sequencing analysis of nematode community composition and structure. Mol Ecol Resour 10:666–676
Prasifka JR, Heinz KM, Winemiller KO (2004) Crop colonization, feeding, and reproduction by the predatory beetle, Hippodamia convergens, as indicated by stable carbon isotope analysis. Ecol Entomol 29:226–233
Putman WL (1965) Paper chromatography to detect predation on mites. Can Entomol 97:435–441
Qiu X, Wu L, Huang H et al (2001) Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl Environ Microbiol 67:880–887
Quéméré R, Hibert F, Miquel C, Lhuillier E, Rasolondraibe et al (2013) A DNA metabarcoding study of a primate dietary diversity and plasticity across its entire fragmented range. PLoS ONE 8:e58971
Quince C, Lanzen A, Davenport RJ et al (2011) Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12:38
Ragsdale DW, Larson AD, Newsom LD (1981) Quantitative assessment of the predators of Nezara viridula eggs and nymphs within a soybean agroecosystem using an ELISA. Environ Entomol 10:402–405
Raso L, Sint D, Mayer R, Plangg S, Recheis T, Brunner S, Kaufmann R, Traugott M (2014) Intraguild predation in pioneer predator communities of alpine glacier forelands. Mol Ecol 23(15):3744–3754
Ratnasingham S, Hebert PDN (2007) BOLD: the barcode of life data system (http://www.barcodinglife.org). Mol Ecol 7:355-364
Rayé G, Miquel C, Coissac E, Redjadj C, Loison A, Taberlet P (2011) New insights on diet variability revealed by DNA barcoding and high-throughput pyrosequencing: chamois diet in autumn as a case study. Ecol Res 26(2):265–276
Reeder J, Knight R (2010) Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods 7:668
Riaz T, Shehzad W, Viari A et al (2011) ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res 39:e145
Richardson RT, Bengtsson-Palme J, Johnson RM (2017) Evaluating and optimizing the performance of software commonly used for the taxonomic classification of DNA metabarcoding sequence data. Mol Ecol Resour 17:760–769
Robasky K, Lewis NE, Church GM (2014) The role of replicates for error mitigation in next-generation sequencing. Nat Rev Genet 15(1):56–62
Rooney N, McCann K, Gellner G, Moore JC (2006) Structural asymmetry and the stability of diverse food webs. Nature 442:265–269
Rothschild G (1966) A study of a natural population of Conomelus anceps Germar (Homoptera: Delphacidae) including observations on predation using the precipitin test. J Anim Ecol 35:413434
Royle JA, Link WA (2006) Generalised site occupancy models allowing for false positive and false negative errors. Ecology 87:835–841
Rubinoff D, Cameron S, Will K (2006) A genomic perspective on the shortcomings of mitochondrial DNA for “barcoding” identification. J Hered 97(6):581–94
Ruess L, Häggblom MM, Zapata EJG, Dighton J (2002) Fatty acids of fungi and nematodes - possible biomarkers in the soil food chain? Soil Biology and Biochemistry 34:745e756
Sanger F, Coulson AR (1975) A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 94(3):441–448
Saqib HSA, Liang P, You M, Gurr GM (2021) Molecular gut content analysis indicates the inter-and intra-guild predation patterns of spiders in conventionally managed vegetable fields. Ecol Evol 11(14):9543–9552
Sarmashghi S, Bohmann K, Gilbert MTP, Bafna V, Mirarab S (2019) Skmer: assembly-free and alignment-free sample identification using genome skims. Genome Biol 20(1):1–20
Schloss PD, Westcott SL, Ryabin T et al (2009) Introducing MOTHUR: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6:e27310
Schmidt BR, Kéry M, Ursenbacher S, Hyman OJ, Collins JP (2013) Site occupancy models in the analysis of environmental DNA presence/absence surveys: a case study of an emerging amphibian pathogen. Methods Ecol Evol 4:646–653
Schnell IB, Bohmann K, Gilbert MTP (2015) Tag jumps illuminated–reducing sequence-to-sample misidentifications in metabarcoding studies. Mol Ecol Resour 15(6):1289–1303
Sims D, Sudbery I, Ilott N et al (2014) Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 15:121–132
Singer GAC, Fahner NA, Barnes JG, McCarthy A, Hajibabaei M (2019) Comprehensive biodiversity analysis via ultra-deep patterned flow cell technology: a case study of eDNA metabarcoding seawater. Sci Rep 9(1):1–12
Shehzad W, Riaz T, Nawaz MA et al (2012) Carnivore diet analysis based on next-generation sequencing: application to the leopard cat (Prionailurus bengalensis) in Pakistan. Mol Ecol 21:1951–1965
Sheppard SK, Bell J, Sunderland KD, Fenlon J, Skervin D, Symondson WO (2005) Detection of secondary predation by PCR analyses of the gut contents of invertebrate generalist predators. Mol Ecol 14(14):4461–4468
Sheppard SK, Harwood JD (2005) Advances in molecular ecology: tracking trophic links through predator–prey food-webs. Funct Ecol 19:751–762
Sigsgaard L, Greenstone MH, Duffield SJ (2002) Egg cannibalism in Helicoverpa zea on sorghum and pigeonpea. Biocontrol 47:151–165
Simon C, Frati F, Beckenbach A et al (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am 87:651–701
Smith DP, Peay KG (2014) Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS ONE 9:e90234
Sopp PI, Sunderland KD (1989) Some factors affecting the detection period of aphid remains in predators using ELISA. Entomol Exp Appl 51(1):11–20
Sopp et al (1992) An improved quantitative method for estimating invertebrate predation in the field using ELISA. J Appl Ecol 79:295–302
Sousa LL, Silva SM, Xavier R (2019) DNA metabarcoding in diet studies: unveiling ecological aspects in aquatic and terrestrial ecosystems. Environmental DNA 1(3):199–214
Srivathsan A, Sha JCM, Vogler AP et al (2015) Comparing the effectiveness of metagenomics and metabarcoding for diet analysis of a leaf feeding monkey (Pygathrix nemaeus). Mol Ecol Resour 15(2):250–261
Stein ED, Martinez MC, Stiles S, Miller PE, Zakharov EV (2014) Is DNA barcoding actually cheaper and faster than traditional morphological methods: results from a survey of freshwater bioassessment efforts in the United States? Casiraghi M, editor. PLoS ONE 9:e95525
Stouffer DB, Camacho J, Jiang W, Nunes Amaral LA (2007) Evidence for the existence of a robust pattern of prey selection in food webs. Proceedings of the Royal Society b: Biological Sciences 274(1621):1931–1940
Sunderland KD, Chambers RJ, Stacey DL, Crook NE (1985) Invertebrate polyphagous predators and cereal aphids. Bulletin SROP 8(3):105–114
Sunderland KD, Crook NE, Stacy DL, Fuller BJ (1987) A study of feeding by polyphagous predators on cereal aphids using ELISA and gut dissection. J Appl Ecol 24:907–933
Sunderland KD (1988) Quantitative methods for detecting invertebrate predation occurring in the field. Annals of Applied Biology 112(1):201–224
Sunderland KD (1996) Progress in quantifying predation using antibody techniques. Systematics Association Special 53:419–456
Sword GA (2000) Tasty on the outside, but toxic in the middle: grasshopper regurgitation and host plant-mediated toxicity to a vertebrate predator. Oecologia 128:416–421
Symondson WOC, Liddell JE (1993) The detection of predation by Abax parallelepipedus and Pterostichus madidus (Coleoptera: Carabidae) on Mollusca using a quantitative ELISA. Bull Entomol Res 83(4):641–647
Symondson WOC, Glen DM, Erickson ML, Liddell JE, Langdon CJ (2000) Do earthworms help to sustain the slug predator Pterostichus melanarius (Coleoptera: Carabidae) within crops? Investigations using a monoclonal antibody-based detection system. Mol Ecol 9:1279–1292
Symondson WOC, Erickson ML, Liddell JE, Jayawardena KGI (1999) Amplified detection, using a monoclonal antibody, of an aphid-specific epitope exposed during digestion in the gut of a predator. Insect Biochem Mol Biol 29(10):873–882
Symondson WOC (2002) Molecular identification of prey in predator diets. Mol Ecol 11:627–641
Sze MA, Schloss PD (2019) The impact of DNA polymerase and number of rounds of amplification in PCR on 16S rRNA gene sequence data. mSphere 4(3):e00163-19
Taberlet P, Coissac E, Pompanon F, Brochmann C, Willerslev E (2012) Towards next‐generation biodiversity assessment using DNA metabarcoding. Mol Ecol 21(8):2045–2050. https://doi.org/10.1111/j.1365-294X.2012.05470.x
Taberlet P., Bonin A., Zinger L., & Coissac E. (2018). Environmental DNA: For biodiversity research and monitoring. Oxford University Press
Tang M, Tan M, Meng G, Yang S, Su XU, Liu S et al (2014) Multiplex sequencing of pooled mitochondrial genomes-a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res 42(22):e166
Thomas AC, Jarman SN, Haman KH, Trites AW, Deagle BE (2014) Improving accuracy of DNA diet estimates using food tissue control materials and an evaluation of proxies for digestion bias. Mol Ecol 23:3706–3718
Thomas AC, Deagle BE, Eveson JP, Harsch CH, Trites AW (2016) Quantitative DNA metabarcoding: improved estimates of species proportional biomass using correction factors derived from control material. Mol Ecol Resour 16(3):714–726
Torr SJ, Wilson PJ, Schofield S et al (2001) Application of DNA markers to identify the individual-specific hosts of tsetse feeding on cattle. Med Vet Entomol 15:78–86
Traugott M, Kamenova S, Ruess L, Seeber J, Plantegenest M (2013) Empirically characterizing trophic networks: what emerging DNA-based methods, stable isotope and fatty acid analyses can offer. Adv Ecol Res 49:177–224
Valentini A, Miquel C, Nawaz MA et al (2009a) New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach. Mol Ecol Resour 9:51–60
Valentini A, Pompanon F, Taberlet P (2009b) DNA barcoding for ecologists. Trends Ecol Evol 24:110–117
van Lenteren J. C., Cock M. J., Hoffmeister T. S., & Sands D. P. (2006). Host specificity in arthropod biological control, methods for testing and interpretation of the data. Environmental impact of invertebrates for biological control of arthropods. Methods and risk assessment. CABI Publishing, Wallingford, UK, 38–63
van der Valk T, Vezzi F, Ormestad M, Dalén L, Guschanski K (2020) Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies. Mol Ecol Resour 20(5):1171–1181
Vance-Chalcraft HD, Rosenheim JA, Vonesh JR, Osenberg CW, Sih A (2007) The influence of intraguild predation on prey suppression and prey release: a meta-analysis. Ecology 88(11):2689–2696
Varennes YD, Boyer S, Wratten SD (2014) Un-nesting DNA Russian dolls - the potential for constructing food webs using residual DNA in empty aphid mummies. Mol Ecol 23(15):3925–3933
Vestheim H, Jarman SN (2008) Blocking primers to enhance PCR amplification of rare sequences in mixed samples - a case study on prey DNA in Antarctic krill stomachs. Front Zool 5:12
Waldner T, Traugott M (2012) DNA-based analysis of regurgitates: a noninvasive approach to examine the diet of invertebrate consumers. Mol Ecol Resour 12(4):669–675
Watts DJ, Strogatz SH (1998) Collective dynamics of ‘small-world’ networks. Nature 393(6684):440–442
Weber DC, Lundgren JG (2009) Quantification of predation using qPCR: effect of prey quantity, elapsed time, chaser diet, and sample preservation. J Insect Sci 9:41
Weber DC, Lundgren JG (2011) Effect of prior diet on consumption and digestion of prey and non-prey food by adults of the generalist predator Coleomegilla maculata. Entomol Exp Appl 140(2):146–152
Willerslev E, Davison J, Moora M et al (2014) Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506:47–51
Williams RJ, Berlow EL, Dunne JA, Barabási AL, Martinez ND (2002) Two degrees of separation in complex food webs. Proc Natl Acad Sci 99(20):12913–12916
Yu DW, Ji Y, Emerson BC et al (2012) Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods Ecol Evol 3:613–623
Zaidi RH, Jaal Z, Hawkes NJ, Hemingway J, Symondson WO (1999) Can multiple-copy sequences of prey DNA be detected amongst the gut contents of invertebrate predators? Mol Ecol 8(12):2081–2087
Zhang GF, Lü ZC, Wan FH, Lövei GL (2007) Real-time PCR quantification of Bemisia tabaci (Homoptera: Aleyrodidae) B-biotype remains in predator guts. Mol Ecol Notes 7:947–954
Zhou X, Li Y, Liu S, Yang Q, Su XU, Zhou L., ... & Huang Q. (2013). Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience, 2(1), 2047-217X
Zinger L, Taberlet P, Schimann H, Bonin A, Boyer F, De Barba M, ... & Chave J (2019). Body size determines soil community assembly in a tropical forest. Molecular Ecology, 28(3), 528-543
Author information
Authors and Affiliations
Contributions
Conception of the work: D. P. P.
Literature search: D. P. P. and D. A. A.
Data analysis: D. A. A.
Draft version: D. P. P. and D. A. A.
Critical revision: D. P. P. and D. A. A.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Edited by Marcos R de Faria.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Paula, D.P., Andow, D.A. DNA High-Throughput Sequencing for Arthropod Gut Content Analysis to Evaluate Effectiveness and Safety of Biological Control Agents. Neotrop Entomol 52, 302–332 (2023). https://doi.org/10.1007/s13744-022-01011-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13744-022-01011-3