
Abstract
This study reports on the preparation and characterization of porous clay heterostructure (PCH) as a high-surface area support for CuO nanoparticles (NPs)-based photocatalyst for the preferential oxidation of methylene blue under visible light in the absence of H2O2. CuO NPs were incorporated in PCH by thermal decomposition method. The new photocatalyst was characterized by DRS, XRD, FT-IR, nitrogen adsorption–desorption measurements and TEM. The results revealed that PCH acts as a host material for spherical CuO NPs with particles size less than 10 nm. The powder X-ray diffraction indicated that PCH contained both MMT clay and MCM-41 and MCM-41 located at the interlayers space of MMT clay. The DRS demonstrated the presence of CuO, Cu2O and Cu NPs in PCH. The synthesized material (CuO–PCH) was a visible light-driven photocatalyst for degradation of MB dye. The photocatalyst demonstrated 94% removal efficiency for MB dye under visible light after 60 min. Kinetics studies showed that the reaction rate constants of CuO–PCH were approximately 1.8, 2.4 and 3.4 times higher than the apparent reaction rate constant of CuO–MMT, CuO–MCM-41 and CuO, respectively. The mechanism of separation of the photogenerated electrons and holes of the CuO–PCH nanocomposite was discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Organic dyes are common pollutants in the effluents from textile, food and cosmetic industries. It is harmful for human beings and toxic to microorganisms [1, 2]. Furthermore, it also has high mobility in water, and can cause great harm to environment and human health. Therefore, the degradation and removal of dyes have gained increasing concern in the past few years. Several approaches have been developed to remove organic dyes from wastewater industries [3,4,5]. One of the increasing ways of reducing aqueous organic dyes is metal oxide semiconductor-assisted photocatalytic process, since it has outstanding features such as low cost, direct use of free solar energy, reusability and it does not release other undesirable chemical [6, 7].
Copper oxide (CuO) has been studied as a p-type semiconductor material with narrow band gap because of the natural abundance of its starting material, low cost of production processing, nontoxic nature, and its reasonably good electrical and optical properties. CuO and Cu2O are p-type semiconductors with narrow band gaps of 1.3–1.7 eV (CuO) and 2–2.5 eV (Cu2O), and they are promising materials with numerous applications in catalyzers, gas sensors, and magnetic materials [8]. However, the CuO exhibited low photocatalytic activity due to fast recombination of photogenerated electron–hole pairs [9]. The binding of CuO with other materials, such as ZnO or TiO2 or adding H2O2 can reduce the recombination of photogenerated electrons–holes, effectively [10, 11].
Since the discovery of the Mobil Catalytic Material (MCMs) in 1992 [12], a lot of research has been conducted in mesoporous materials synthesized by a template mechanism involving the polymerization of a silica source around surfactant molecules [13]. Pure siliceous MCM-41 material possess many unique properties such as high surface area, high pore volume, as well as parallel and ideally shaped pore structures without the complications of a network [14]. In 1998, Pinnavaia [15] applied the MCM and pillared clay preparation technologies on natural clays, obtaining a new interesting large-pore derivative, designated porous clays heterostructure or briefly PCH. For this new solid, the silica source has been polymerized in situ between the aluminosilicate sheets and around micellar rods of surfactant and co-surfactant previously ion-exchanged on the interlayer space of the natural clay. After calcination for the removal of the organics, the mesopores are formed and the PCH obtained. Characteristics common to the PCH materials are their high surface area (250–1000 m2 g−1) and a combination of micro- and mesoporosity. Also, in comparison to MCMs they have important advantages such as their good thermal stability and mechanical strength [16].
In our previous studies, incorporation of copper oxide was investigated in MCM-41 mesoporous material [17], MMT clay [18] and magnetic (Fe3O4) core [4] matrices in order to degradation of organic dye or researching antibacterial activity CuO. Recently, Aguiar et al. [19] used PCH from natural bentonite for adsorption of the dye molecules. Cecilin et al. reported [20] the preparation and characterization of porous clay heterostructures (PCH) as a high-surface-area support for CuO–CeO2-based catalysts for the preferential oxidation of CO in excess of H2 (CO-PROX). To the best of our knowledge, there is no report on the controlled synthesis of CuO–PCH as a visible light-driven photocatalyst. Herein, in this study, the montmorillonite-based PCH material, intercalated with the pure MCM-41was constructed. The PCH with high surface area was used as a support for incorporation of CuO NPs by thermal decomposition method. The new material was applied for degradation of methylene blue (MB) dye in the absence H2O2.
Experimental
Materials
The commercial sodium montmorillonite clay (NaMMT), < 25 μm montmorillonite, cation exchange capacity (CEC = 92.6 meq/100 g) was purchased from Southern Clay Products (Gonzales, TX, USA), Na2CO3, CuSO4.5H2O and ethanol were purchased from Merck, Germany. Tetraethylorthosilicate (TEOS, 98%), ammonia (25 wt%), the surfactant cetyltrimethylammonium bromide (CTABr), and dodecylamine (DDA) were purchased from Fluka and Merck.
Synthesis of PCH
The 1.5 g NaMMT was added to 50 mL solution of 0.1 M surfactant (CTABr) and stirred at 60 °C for 24 h. Then, the (CTAB-MMT) was separated from the solution and washed with demineralized water and ethanol till pH of 7 was reached. The sample was dried at 60 °C for 3 h. 0.723 g of dried CTABr-MMT was stirred in 11.465 g dodecylamine (as co-surfactant) for 30 min at 50 °C. Subsequently, the tetraethylorthosilicate was added into suspension and was allowed to react for 4 h under continuous stirring at room temperature. The following molar ratio of CTABr/DDA/TEOS was applied: 1/20/150 for the production of the sample, which, respectively, is denoted as PCH. After that the modified MMT were separated from the solutions, washed with pure ethanol, dried at room temperature and finally calcined at temperatures 600 °C for 5 h.
Synthesis of CuO–PCH photocatalyst
CuO NPs were incorporated in PCH via direct thermal decomposition method. For preparation of CuO–PCH 0.4 g Na2CO3 and 0.3 g CuSO4.5H2O was added to 20 ml deionized water and was stirred at 60 °C. After 10 min, 0.5 g PCH was added to suspension. The mixture was stirred for forming green participate, Cu4 (SO4) (OH)6–PCH was separated by filtration and washed several times with warm deionized water to remove possible remaining ions in the final product. Resultant Cu4 (SO4) (OH)6–PCH was transferred to a silica crucible and placed in an oven at 70 °C for 6–7 h. Finally, it was kept in a pre-heated muffle furnace at 600 °C for decomposition. After 5 h, the silica crucible was taken out from the furnace and allowed to cool to room temperature following which the collected mass was ground. The resultant brownish black powder was collected. The reaction pathway of thermal decomposition of Cu4 (SO4)(OH)6-PCH to CuO–PCH NPs is represented by the following reactions:
The prepared sample is termed as CuO–PCH. For preparing CuO NPs in MMT or MCM-41only above procedure was used; MCM-41 or MMT was added instead of PCH. CuO NPs were prepared via direct thermal decomposition method without adding support.
Evaluation of photocatalytic activity
Photodegradation experiments were performed with a photocatalytic reactor system. This bench-scale system consisted of cylindrical Pyrex-glass cell with 250 mL capacity, 10 cm inside diameter and 15 cm height. A 200 W tungsten filament Philips lamp (λ > 400 nm) was placed in a 5-cm-diameter quartz tube with one end tightly sealed by a Teflon stop the lamp and the tube were then immersed in the photoreactor cell with a light path of 3 cm. The photoreactor was filled with 100 ml of 3.2–16 ppm of dye as pollutant and 0.03–0.1 g l−1 of photocatalysts. The whole reactor was cooled with water-cooled jacket on its outside and the temperature was kept at 25 °C. Before the suspensions were irradiated, they were magnetically stirred for 30 min in the dark to complete the adsorption–desorption equilibrium between the dye and the surface of the photocatalysts. Lastly, the quartz reactor containing suspensions were exposed to visible light irradiation under stirring. All reactants in the reactions were stirred using a magnetic stirrer to ensure that the suspension of the catalyst was uniform during the course of the reaction. To determine the percent of the destruction of dyes, the samples were collected at regular intervals, and centrifuged to remove the photocatalyst particles that exist as undissolved particles in the samples.
A separate control experiment was also performed to observe MB decolorization and degradation in the absence of photocatalysts (photolysis). The photocatalytic activity was evaluated in terms of decolorization (oxidation of chromophore group) and degradation (oxidation of aromatic group) efficiencies. The decolorization and degradation efficiencies were calculated with respect to the change in intensities of absorption peaks at 664 nm (λmax of MB) and 280 nm (representative λ for aromatic carbon content) using the following equation:
where A0 is the equilibrium absorbance at 664 and 280 nm after adsorption–desorption equilibrium and At is the absorbance at 664 and 280 nm after a certain irradiation time (min). The absorption of the solution was measured by a UV–Vis spectrophotometer Shimadzu model 1600 PC. In order to obtain maximum degradation efficiency, concentration of the dye, pH and the amount of core–shell in photocatalyst were studied in amplitudes of 3.2–16 ppm, 3–11 and 0.03–0.1 g l−1, respectively.
Characterization
The powder X-ray diffraction patterns of the samples were recorded using an X-ray diffractometer (X Pert Prompd) with Cu Kα radiation (λ = 1.545 Å) under the conditions of 45 KV and 30 mA, at a step size of 2θ = 0.02°. X-ray diffraction (XRD) patterns were recorded between 2° and 70° 2θ at a scanning speed of 2° min−1. All samples were analyzed in random orientation. The transmission electron micrographs (TEM) were recorded with a Philips CM10 microscope, working at a 100 kV accelerating voltage. Samples for TEM were prepared by dispersing the powdered sample in acetone by sonication and then drip drying on a copper grid coated with carbon film. Samples were sonicated for 15 min. Scanning electron microscope (SEM) was conducted with a TESCAN MIRA3 scanning electron microscope operated at 30 kV. The specific surface area and pore diameter were measured using a Sibata Surface Area Apparatus 1100. The infrared spectra were measured on a Bruck spectrophotometer using KBr pellets.
Results and discussion
Characterization of PCH and CuO–PCH photocatalyst
XRD analysis
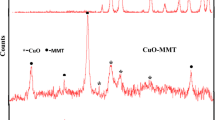
The crystalline structure of the MMT, PCH and CuO–PCH was investigated by XRD, and the corresponding patterns are illustrated in Fig. 1. The XRD patterns of pure MMT and pure CuO were also presented as controls. The diffraction peaks for CuO can be indexed by the characteristic 32.67° (111), 35.63° (002), 38.90° (− 111), 48.83° (− 202), 53.60° (020), 58.24° (202), 61.59° (− 113), 66.5° (022) and 68.19° (220) reflections of monoclinic phase of CuO (with S.G. C2/c; and lattice constants of a = 4.6893 Å, b = 3.4268 Å, c = 5.1321 Å and beta = 99.6530°) which match very well with JCPDS 05-0661 [21]. For MMT the diffraction peak d001 attributed to the ordering of the MMT layers is present in the position (7.235° 2θ) related to the basal spacing of 1.22 nm and two-dimensional diffractions h k was found at 19.91 and 35.20 [22]. Additionally, the reflection at 2θ of 19.91° corresponding to diffraction from (100) layers of montmorillonite were detected. The basal spacing of the layered clay minerals depends on the thickness of the clay layer and interlayer distance. The thickness of the montmorillonite layers is estimated to be about 0.96 nm [23, 24]. Therefore, the interlayer distance is about 0.26 nm, which is typical of hydrated clays [25]. The h k reflections are characteristic of this type of the clay mineral. Each observed h k reflection is the summation of several h k index pairs. The diffraction at 19.91° is the summation of h k indices of (02) and (11), while the diffraction at 35.20 is superposition of (13) and (20) indices [23]. The XRD pattern of PCH showed peaks that could be indexed to MCM-41 mesoporous structure and MMT [2]. The diffraction pattern of CuO–PCH photocatalyst indicated the reflections at 2θ = 35.55° (002), 38.85° (− 111), and 48.75° (− 202) that correspond to the peaks of copper oxide and other reflections that could be related to MCM-41 and MMT. The deposition of surfactants and co-surfactants as well as the formation of the CuO NPs in the interlayer space of montmorillonite resulted in a shift of the d001 peak in the direction of the lower values of 2θ angles. This effect is related to an increase of the interlayer distance in the clay materials. For the PCH and CuO–PCH samples, the 001 peaks were shifted to the position characteristic for the basal spacing of 3.85 and 2.96 nm, respectively. Taking into account the thickness of the montmorillonite layer (0.96 nm), it could be calculated that the interlayer distance is about 2.89 and 2.00 nm, respectively. There was broadening of CuO–PCH peaks in comparison with PCH alone, and this can be related to the presence of copper oxide.

XRD patterns of samples
N2 sorption–desorption isotherms
The nitrogen sorption–desorption isotherms as well as BJH pore size distributions for PCH and CuO–PCH samples are presented in Fig. 2. A gradual increase in nitrogen sorption observed at low-to-medium partial pressure (p/po < 0.3) suggests the presence of supermicropores and small mesopores (Fig. 2a) [26]. An increase in adsorbed volume observed at higher partial pressures could be related to larger mesopores. The hysteresis loops could be qualified to the H4 type, corresponding to the aggregates forming the slit-like pores with uniform sizes [27]. Figure 2b shows that pore diameters of sample was ~ 2.48 nm in samples. The BET surface areas (SBET) and pore diameters (Table 1) were calculated using the BET method. Both PCH and CuO–PCH show a rise of the SBET from 77 m2 g−1 for the bare montmorillonite to 905 for PCH and 875 m2 g−1 for CuO–PCH by the insertion of pillars between the 2:1 layers of montmorillonite. The introduction of the copper oxide system to the PCH produces a decrease in surface area and cumulative pore volume due to a partial blockage, mainly of the microporous framework (Table 1). Despite these textural parameters decrease with the increasing of CuO-loading, CuO supported on PCH exhibits a higher surface area than the bulk CuO system [21], favoring the presence of a higher amount of active centers with a noteworthy dispersion as shown by the XRD pattern (Fig. 1).

a Adsorption–desorption isotherms and b BJH pore size distribution of the PCH and CuO–PCH samples
FT-IR investigations
The FT-IR spectra of PCH and CuO–PCH samples are shown in Fig. 3A. PCH sample showed the most intensive band at 1048 cm−1 which was attributed to Si–O in-plane stretching vibration. The band at 450 cm−1 was due to Si–O–Si bending vibrations and 775 cm−1 was attributed to FeMg–OH bending vibration. The OH-stretching band at 3441 cm−1 was due to hydroxyl group bonded with Al3+ cations [28]. The broadness of 3441 cm−1 band showed the substitution of octahedral Al3+ by Fe3+ or Mg2+ cations. Two bending vibrations of hydroxyl groups associated with these cations, i.e., 935 cm−1 (AlAlOH), and 775 cm−1 (AlMgOH) confirm the substitution in octahedral layer. The presence of Al3+, Fe3+ and Mg2+ cations in PCH structure were attributed to MMT cations in its structure. Most bands in the CuO–PCH sample showed a shift to lower wave numbers and intensity decreased compared to the PCH matrix. This confirmed the presence of nanosized CuO particles present in the PCH matrix. Vaseem et al. studied structural and photocatalytic characteristics of CuO nanostructures and concluded that the intensity increase of band located at 446 cm−1 belong to the monoclinic phase of CuO [29].

a FT-IR spectra of PCH and CuO–PCH and b UV–Vis diffuse reflectance spectra of PCH, CuO–PCH and CuO nanoparticles (insert)
Study of UV–Vis diffuse reflectance spectra of nanocatalyst
Figure 3b presents the UV–Vis diffuse reflectance spectra (DRS) recorded for the PCH and CuO–PCH samples calcined. The PCH sample contained iron (FT-IR spectrum), which is a natural component of montmorillonite. The DR spectra of the iron containing samples are characterized by the bands related to Fe3+ ← O charge transfer. The position of these bands depends on the coordination and agglomeration of iron (III) species [30]. Mononuclear Fe3+ cations give rise to the bands in the range of 200–300 nm. The bands in the region of 300–400 nm are characteristic of small oligonuclear FexOy clusters, while bulky particles of Fe2O3 give characteristic bands above 400 nm [31]. Additionally, in the case of isolated Fe3+ cations a distinction between iron ions in the tetrahedral (band below 250 nm) and octahedral (band in the range of 250–300 nm) coordination is possible [30]. The spectrum recorded for the PCH sample showed a maximum band in the range of 250–300 nm that is related to the isolated Fe3+ cations located in the octahedral positions of the clay layers. In the spectrum of CuO nanoparticles (insert of Fig. 3b), the band at 270 nm is shown, which can be attributed the due to O2− to Cu2+ charge-transfer transitions [32]. The broad band observed in the region between 350 and 700 nm in the spectra of CuO nanoparticles. This band can be assigned to the d–d transition of Cu with octahedral environment in CuO [33]. The DR spectrum of CuO–PCH nanocatalyst (Fig. 3b) showed broad bands around 340–360, 400–460, 480–550 and 700–800 nm corresponding to the presence of [Cu–O–Cu]n clusters, Cu2O, Cu nanoparticles and CuO samples, respectively [34]. The band gap energy (Eg) of Cu2O and CuO nanoparticles in CuO–PCH nanocatalyst was estimated from the Eg = hc/λ (eV) equation. The calculated Eg value of Cu2O and CuO was ~ 2.6 and 1.53 eV, respectively, which was in agreement with other articles [21]. The result also demonstrated that the CuO–PCH nanocatalyst could sufficiently absorb visible light and possess favorable photocatalytic properties under visible light irradiation.
The conduction and valence band positions in the semiconductors at the point of zero charge can be calculated by the following formula [35]:
where \({E_{{V_{\text{B}}}}}\) is the potential of the valence band, X is the electronegativity of the semiconductor which was the geometric mean of the electronegativity of the constituent atoms. The electronegativity value for CuO was 5.81 eV [35]. The Ee was the energy of free electrons on the hydrogen scale (4.5 eV) and Eg was the band gap energy of the semiconductor. In the present study, the band gap energy of the CuO in CuO–PCH nanocatalyst was 1.53 eV. The \({E_{{V_{\text{B}}}}}\) and \({E_{{C_{\text{B}}}}}\) were calculated as 2.07 and 0.54 eV, respectively. Similarly, the \({E_{{V_{\text{B}}}}}\) and \({E_{{C_{\text{B}}}}}\) of Cu2O in CuO–PCH nanocatalyst were calculated as 2.12 and − 0.48 eV, respectively.
Study of TEM images
TEM images were used to determine the morphology of the synthesized materials and estimate the size of CuO nanoparticles in CuO–PCH sample. The TEM analysis results (Fig. 4) showed that the original PCH had an obvious layered structure, whereas many small spherical CuO nanoparticles were incorporated into the surface of the PCH. The mean diameter of CuO nanoparticles was evaluated at range of 5–10 nm.

TEM images of PCH and CuO–PCH samples
Effect of photocatalytic experimental parameters
The effect of CuO–PCH photocatalyst dosage on the photodegradation of MB was studied in the range of 0.03–0.1 g l−1. The initial rates of the degradation was enhanced with increasing the amount of the photocatalyst up to an optimum weight of CuO–PCH (0.08 g l−1) and further increase had a negative effect. Increasing the amount of the photocatalyst, increases the number of adsorbed MB molecules onto the photocatalyst surface, but excess photocatalyst particles in the solution, decreases the light penetration and hence reduces the photodegradation rate [36]. Another reason was the aggregation of solid particles when a large amount of photocatalyst was used [37].
The effect of initial concentration of the solution on the photocatalytic degradation rate was investigated over the concentration range of 3.2–16 ppm MB. The degradation efficiency of dye decreased with increasing the initial concentration of dye to more than 10 ppm. At higher concentration of MB in the solution, the photons would be absorbed by the MB molecules before they could reach photocatalyst surface. This in turn reduces absorption of photons by the photocatalyst and consequently the degradation efficiency [37].
The pH of the solution affects the photodegradation processes due to the strong pH dependence of the properties such as semiconductor’s surface charge state, flat band potential, and dissociation of compounds in the solution. The effect of pH was investigated on the efficiency of the photodegradation process under optimal conditions in the pH range of 3–11. In the presence of 0.08 g l−1 CuO–PCH sample with increasing pH from 3 to 9, degradation efficiency rose from 20 to 60%, respectively, and decreased to 42% at pH 11 for 60 min of irradiation. The pH of point of zero charge (pHpzc) calculated for the CuO NPs by different methods and reported between 8.5 and 9.5 [35]. The value of pHPZC for synthesized photocatalyst was calculated to be 8.4. The charge of the photocatalyst surface was positive and negative at pH values smaller and higher than the pHpzc, respectively. Hence, at pH values smaller than pHpzc, the repulsion between the positively charged surface of the CuO–PCH photocatalyst and positive groups of MB molecules decreases the photodegradation efficiency. When pH > 8.4 the surface of the catalyst was slightly negatively charged, and the coordinative interaction with positive R2 = N+ = R/ group of MB is increased.
Photocatalytic activity evaluation
As is well known, the effective adsorption of dyes on the surface of the photocatalyst is the precondition of photocatalytic degradation [33]. The photocatalytic activity of CuO–PCH photocatalyst for MB was assessed by performing experiments with 10 ppm aqueous MB solutions at a catalyst dosage of 0.08 g l−1 under visible light irradiation. The reaction pH was controlled during the photodegradation experiments conducted in 9 for 60 min. The photocatalytic performance of the resultant photocatalysts was evaluated in λ = 664 and 280 nm by photodegradation and photodecolorization of MB as a contaminant under visible light irradiation. In the absence of photocatalyst, MB self-degradation was almost negligible. Figure 5a shows the percent degradation curves of MB dye over CuO–MCM-41, CuO–MMT, CuO NPs, and CuO–PCH photocatalyst. It was found that the direct visible light photolysis resulted in only about 5% degradation in 60 min accompanied with no degradation in the absence of CuO–PCH photocatalyst. The above results implied that visible light photolysis was not sufficient in the decolorization of MB aqueous solution and degradation of its organic carbon content. CuO–MCM-41, CuO–MMT, PCH and CuO samples showed ~ 15, 22, 20 and 18% degradation of the MB dye after 60 min. However, as can be followed from Fig. 5b, MB aqueous solution decolorized and degraded efficiently in the presence of CuO–PCH photocatalyst by visible light irradiation. The characteristic absorption peak of MB at 664 nm gradually decreased during the photocatalytic oxidation experiments indicating the cleavage of the chromophore structure of MB. The abatement of organic carbon content of MB was also evident from the decrement of absorbance at 280 nm. According to Fig. 5b, 59 and 60% degradation and decolorization (or the removal of MB dye ~ 94%) were achieved for CuO–PCH photocatalyst after 60 min photocatalysis. Considering the above findings, the degradation rate of the MB dye in the presence of CuO–PCH photocatalyst is similar to achieve decolorization. The CuO NPs due to low surface area indicated low degradation for MB dye. The PCH photocatalyst due to high surface area showed high adsorption in the dark time, but in the presence of visible light, degradation was very low because there was no semiconductor in PCH. In our previous work, CuO–MMT [18] showed 60% decolorization for MB dye, but low degradation (~ 22%) was observed in the absence of H2O2. The CuO–MCM-41 with 870 m2g−1 showed only ~ 15% degradation for the MB dye. Generally, the valance band potential of CuO is not negative [38] enough compared with the standard reduction potential of ·OH/H2O [39], indicating that the photogenerated hole (h+) on the surface of CuO cannot oxidize H2O into ·OH and photocatalysis process needs H2O2 solution. The degradation of MB in the presence of CuO–PCH without H2O2 solution was ~ 59%. Probably, the introduction of PCH plays an active part in the adsorption and catalytic degradation process of the MB dye. Two reasons may account for the enhanced photocatalytic activity of the CuO–PCH photocatalyst in the absence of H2O2: (1) PCH support can adsorb more MB dye molecules on its surface (owing to high surface area); (2) due to existence mesoporous pore and aluminum atoms in PCH structure, probably, PCH can transfer electron allowing for the photo-excited electrons of CuO in the photocatalyst to be quickly transferred from the conduction band of CuO to PCH (reducing recombination of electron–hole in CuO). Subsequently, the electrons can be captured by the adsorbed O2, resulting in the formation of highly reactive hydroxyl radicals (·OH) or (·O2−) for degradation of organic pollutants.

a Degradation percent of different photocatalysts for MB aqueous solution and b absorption spectra changes of a solution of MB in the presence of CuO–PCH photocatalyst, under visible light, Co MB = 10 ppm, amount photocatalysts = 0.08 g l−1, pH 9 and in irradiation time 60 min
Photocatalytic stability of samples
The photostability of a photocatalyst is a very important characteristic parameter with respect to the practical application. To study the photostability of the synthesized CuO–PCH photocatalyst, five photocatalytic experimental runs were performed by adding used photocatalyst to fresh MB solutions with no change in overall concentration of the catalyst under visible light irradiation. In each recycling run, the photocatalyst was reused after centrifuging, washed with double distilled water and dried at 80 °C while other factors were kept identical. The stability of new photocatalyst is displayed in Fig. 6a. The photocatalytic activity of the CuO–PCH photocatalyst was retained at ~ 88% the removal of MB dye even after five successive experimental runs under the same conditions, which elevates the photocatalyst for its practical applications in environmental remediation. Therefore, CuO–PCH photocatalyst indicated a higher stability with respect to CuO NPs [24] and other nanocomposites such as, Fe3O4@CuO [40], Fe3O4@CuO-RGO core–shell [41].

a The cycling removal efficiency of MB solution in the presence of CuO–PCH photocatalyst. b Ln(C0/C) versus irradiation time plot
Photocatalytic reaction kinetics can be expressed by the pseudo-first-order model, Ln (C0/C) = kappt, which is obtained by simplification of Langmuir–Hinshelwood (L–H) kinetics model [42]; where kapp is the apparent pseudo-first-order reaction rate constant and t is the reaction time. A plot of ln (C0/C) versus t will yield a slope of kapp. The calculated kapp is presented in Fig. 6b. The degradation process follows pseudo-first-order kinetics and the degradation rate constant calculated from the slope of the kinetics plot was found to be 0.0215, 0.0114, 0.0088 and 0.0062 min−1 for CuO–PCH, Cuo–MMT, CuO–MCM-41 and CuO samples, respectively.
Reaction mechanism
It was reported [39], that in the absence of H2O2, CuO is not an effective photocatalyst for degrading organic pollutants. This result may be attributed to a fact that the VB of CuO is more negative than the redox potential necessary for the generation of radicals similar to the present work (Fig. 7) that the \({E_{{V_{\text{B}}}}}\) and \({E_{{C_{\text{B}}}}}\) were calculated as 2.06 and 0.55, respectively. In addition to CuO NPs, the DRS results demonstrated the presence of Cu2O and Cu nanoparticles in PCH support. The binding of CuO with Cu2O NPs can reduce the recombination of photogenerated electrons–holes, effectively [10, 11]. As shown in Fig. 7, in CuO–PCH photocatalyst both CuO and Cu2O NPs can absorb visible light due to its small band gap (1.53 and 2.6 eV, respectively) to produce electrons and holes. The electrons of Cu2O NPs will be quickly injected into the conduction band of Cu and then CuO NPs, respectively. Normally, when two materials with different work functions are combined with each other, a Schottky barrier will be established and electrons will transfer from the materials with a high work function to the materials with a low work function [43]. From the photochemistry point of view, it was not possible to reduce O2 to ·O2−, E° (O2/·O2−= − 0.28 V/SHE) [44] through one-electron reduction process because the CB potential of CuO (0.54 eV versus SHE) was more positive than the single-electron reduction potential of oxygen. The VB of CuO is more negative than the redox potential necessary for the generation of ·OH radicals (Fig. 7). On the other hand, the photogenerated holes are key activated species for discoloration of MB. Ultimately, the active species (holes, ·OH) oxidized the MB molecules that adsorbed on the active sites of the CuO–PCH via electrostatic attraction, resulting in the complete decomposition of MB into harmless gaseous CO2 and inorganic ions [3].

Schematic illustration for a possible photocatalytic mechanism of the prepared CuO–PCH photocatalyst under visible light irradiation
The formation of Cu2O/Cu/CuO heterojunction ensures a large space-charge-region potential, which significantly improves the electron–hole separation efficiency and lowers the electron–hole recombination rate. In other words, the large space-charge-region potential at Cu2O/Cu/CuO interface facilitates not only the injection of photoinduced electrons from the conduction band of Cu2O to that of CuO, but also the injection of photoinduced holes from the valence band of Cu2O to that of CuO. Finally, the Cu2O/CuO bilayered composite has a greatly improved charge carrier density, and thus ensures a faster carrier transportation rate inside the copper oxides.
Conclusions
The montmotollonite-based PCH material intercalated with CuO nanoparticles was synthesized and characterized. CuO nanoparticles were incorporated in the PCH support by thermal decomposition method. The CuO–PCH nanocomposite was used as photocatalysts to degrade organic pollutants without additional H2O2. The degradation rate of MB was considerably enhanced by CuO loaded on PCH under visible light irradiation. The CuO nanoparticles, CuO–MCM-41, CuO–MMT and PCH did not have significant degradation efficiency. The aluminum atoms and mesoporous material in PCH showed an important role in the degradation process. MCM-41 increased adsorption of MB dye and aluminum atoms in MMT clay reduced recombination of electron–hole in CuO nanoparticles.
References
J. Xie, H. Wang, M. Duan, L. Zhang, Synthesis and photocatalysis properties of ZnO structures with different. Appl. Surf. Sci. 257, 6358–6363 (2011)
A. Jafarzadeh, Sh. Sohrabnezhad, M.A. Zanjanchi, M. Arvand, Synthesis and characterization of thiol-functionalized MCM-41 nanofibers and its application as photocatalyst. Microporous Mesoporous Mater. 236, 109–119 (2016)
Sh. Sohrabnezhad, A. Seifi, The green synthesis of Ag/ZnO in montmorillonite with enhanced photocatalytic activity. Appl. Surf. Sci. 386, 33–40 (2016)
S.K. Rajabi, Sh Sohrabnezhad, S. Ghafourian, Fabrication of Fe3O4@CuO core-shell from MOF based materials and its antibacterial activity. J. Solid State Chem. 244, 160–163 (2016)
J. Zhu, H. Fan, J. Sun, S. Ali, Anion-exchange precipitation synthesis of alfa-Ag2WO4/Zn-Cr layered double hydroxides composite with enhanced visible-light-driven photocatalytic activity. Sep. Purif. Technol. 120, 134–140 (2013)
F. Petronella, A. Truppi, C. Ingrosso, T. Placido, M. Striccoli, M.L. Curri, A. Agostiano, R. Comparelli, Nanocomposite materials for photocatalytic degradation of pollutants. Catal. Today 281, 29–37 (2017)
R. Ameta, R.K. Malkani, A comparative study of photocatalytic activity of some coloured semiconducting oxides. Iran. J. Chem. Chem. Eng. 29, 43–48 (2010)
M. Yin, F. Wang, H. Fan, L. Xu, S. Liu, Heteroj unction CuO@ZnO microcubes for superior p-type gas sensor application. J. Alloys Compd. 672, 374–379 (2016)
R. Gusain, P. Kumar, O.P. Sharma, S.L. Jain, O.P. Khatri, Reduced graphene oxide–CuO nanocomposites for photocatalytic conversion of CO into methanol under visible light irradiation. Appl. Catal. B: Environ. 181, 35 2–362 (2016)
K. Mageshwari, D. Nataraj, T. Pal, R. Sathyamoorthy, J. Park, Improved photocatalytic activity of ZnO coupled CuO nanocomposites synthesized by reflux condensation method. J. Alloys Compd. 625, 362–370 (2015)
B. Fang, Y. Xing, A. Bonakdarpour, S. Zhang, D.P. Wilkinson, Hierarchical CuO-TiO2 hollow microspheres for highly efficient photo-driven reduction of CO2 to CH, ACS Sustainable. Chem. Eng. 3, 2381–2388 (2015)
J.S. Beck, J.C. Vartuli, W.J. Roth, M.E. Leonowicz, C.T. Kresge, K.D. Schmitt, T.-W. Chu, D.H. Olson, E.W. Sheppard, S.B. Mc Cullen, J.B. Higgins, J.L. Schlenker, A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 114, 10834–10843 (1992)
C.T. Kresge, M.E. Leonowicz, W.J. Roth, J.C. Vartuli, J.S. Beck, Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature. 359, 710–712 (1992)
J. Choma, M. Jaroniec, W. Burakiewicz-Mortka, M. Kloske, Critical appraisal of classical methods for determination of mesopore size distributions of MCM-41 materials. Appl. Surf. Sci. 196, 216–223 (2002)
T.J. Pinnavaia, Porous clay heterostructures prepared by gallery templated synthesis, US patent 1998. (US 5834391)
A. Galarneau, A. Barodawalla, T.J. Pinnavaia, A kinase from fission yeast responsible for blocking mitosis in S phase. Nature 374, 529–531 (1995)
S. Sohrabnezhad, A. Valipour, Synthesis of Cu/CuO nanoparticles in mesoporous material by solid state Reaction. Spectrochim. Acta A 114, 298–302 (2013)
S. Sohrabnezhad, A. Pourahmad, T. Salavatiyan, CuO-MMT nanocomposite: effective photocatalyst for the discoloration of methylene blue in the absence of H2O2. Appl. Phys. A 122, 111–123 (2016)
J.E. Aguiar, J.A. Cecilia, P.A.S. Tavares, D.C.S. Azevedo, E. Rodríguez Castellón, S.M.P. Lucena, I.J. Silva Junior, Adsorption study of reactive dyes onto porous clay heterostructures. Appl. Clay Sci 135, 35–44 (2017)
J.A. Cecilia, A. Arango-Díaz, F. Franco, J. Jiménez-Jiménez, L. Storaro, E. Moretti, E. Rodríguez-Castellón, CuO-CeO2 supported on montmorillonite-derived porous clay heterostructures (PCH) for preferential CO oxidation in H2-rich stream. Catal. Today 253, 126–136 (2015)
L. Yang, D. Chu, L. Wang, CuO core-shell nanostructures: precursor-mediated fabrication and visible-light induced photocatalytic degradation of organic pollutants. Powder Technol. 287(3), 46–354 (2016)
R.M. Torres Sanchez, M. Genet, S. Yunes, X.V. Cong. AFQQI. C-95 Tandil, (2007)
G.W. Brindley, R.W. Hoffmann, Orientation and packing of aliphatic chain molecules on montmorillonite, clay organic studies IV. Clays Clay Miner. 9, 546–556 (1962)
S. Xu, S.A. Boyd, Cationic surfactant adsorption by swelling and nonswelling layer silicates. Langmuir 11, 2508–2514 (1995)
M. Kawano, M. Tomita, Dehydration and rehydration of saponite and vermiculite. Clays Clay Miner. 39, 174–183 (1991)
M. Benjelloun, P. Cool, T. Linssen, E.F. Vansant, Acidic porous clay heterostructures: study of their cation exchange capacity. Microporous Mesoporous Mater. 49, 83–94 (2001)
S.J. Greek, K.S.W. Sing, Adsorption, Surface Area and Porosity (Academic, London, 1982)
V.C. Farmer, J.D. Russell, Effects of particle size and structure on the vibrational frequencies of layer silicates. Spectrochim. Acta. A 22, 389–398 (1966)
S.P. Meshram, P.V. Adhyapak, U.P. Mulik, D.P. Amalnerkar, Facile synthesis of CuO nanomorphs and their morphology dependent sunlight driven photocatalytic properties, Chem. Eng. J. 204–206, 158–165 (2012)
M.S. Kumar, M. Schwidder, W. Grunet, A. Bruckner, On the nature of different iron sites and their catalytic role in Fe-ZSM-5 DeNOx catalysts: new insights by a combined EPR and UV/VIS spectroscopic approach. J. Catal. 227, 384–397 (2004)
M.S. Kumar, J. Perez-Ramirez, M.N. Debbagh, S. Smarsly, U. Bentrup, A. Bruckner, Evidence of the vital role of the pore network on various catalytic conversions of N2O over Fe-silicalite and Fe-SBA-15 with the same iron constitution. Appl. Catal. B 62, 244–254 (2006)
A. Nezamzadeh-Ejhieh, M. Karimi-Shamsabadi, Decolorization of a binary azo dyes mixture using CuO incorporated nanozeolite-X as a heterogeneous catalyst and solar irradiation. Chem. Eng. J. 228, 631–641 (2013)
A.A. Jalil, M.A.H. Satar, S. Triwahyono, H.D. Setiabudi, N.H.N. Kamarudin, N.F. Jaafar, N. Sapawe, R. Ahamad, Tailoring the current density to enhance photocatalytic activity of CuO/HY for decolorization of malachite green. J. Electroanal. Chem. 701, 50 (2013)
P. Wang, X. Zheng, X. Wu, X. Wei, L. Zhou, Preparation and characterization of CuO nanoparticles encapsulated in mesoporous Silica. Microporous Mesoporous Mater. 149, 181–185 (2012)
Y. Xu, M.A.A. Schoonen, The absolute energy positions of conduction and valence bands of selected semiconducting minerals. Am. Mineral. 85, 543–556 (2000)
A. Riga, K. Soutsas, K. Ntampegliotis, V. Karayannis, G. Papapolymerou, Effect of system parameters and of inorganic salts on the decolorization and degradation of Procion H-exl dyes. Comparison of H2O2/UV, Fenton, UV/Fenton, TiO2/UV and TiO2/UV/H2O2 processes. Desalination 211, 72 (2007)
C.H. Chiou, C.Y. Wu, R.S. Juang, Influence of operating parameters on photocatalytic degradation of phenol in UV/TiO2 process. Chem. Eng. J. 139, 322 (2008)
Q. Zhang, K. Zhang, D. Xu, G. Yang, H. Huang, F. Nie, Ch Liu, Sh. Yang, CuO nanostructures: synthesis, characterization, growth mechanisms, fundamental properties, and applications. Prog. Mater Sci. 60, 208–337 (2014)
M. Miyauchi, A. Nakajima, T. Watanabe, K. Hashimoto, Photocatalysis and photoinduced hydrophilicity of various metal oxide thin films. Chem. Mater 14, 2812–2816 (2002)
Y.-F. Zhang, L.-G. Qiu, Y.-P. Yuan, Y.-J. Zhu, X. Jiang, J.-D. Xiao, Magnetic Fe3O4@C/Cu and Fe3O4@CuO core–shell composites constructed from MOF-based materials and their photocatalytic properties under visible light. Appl. Catal. B 144, 863–869 (2014)
J. Ding, L. Liu, J. Xue, Zh Zhou, G. He, H. Chen, Low-temperature preparation of magnetically separable Fe3O4@CuO-RGO core-shell heterojunctions for high-performance removal of organic dye under visible light. J. Alloys Compd. 688, 649–656 (2016)
L. Latifi, Sh Sohrabnezhad, M. Hadavi, Mesoporous silica as a support for poorly soluble drug: Influence of pH and amino group on the drug release. Microporous Mesoporous Mater. 250, 148–157 (2017)
X. Zhang, Y.L. Chen, R.Sh. Liu, D.P. Tsai, Plasmonic photocatalysis. Rep. Prog.Phys. 76, 046401–046442 (2013)
Y. Liang, N. Guo, L. Li, R. Li, G. Ji, S. Gan, Fabrication of porous 3D flower-like Ag/ZnO heterostructure composites with enhanced photocatalytic performance. Appl. Surf. Sci. 332, 32–39 (2015)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sohrabnezhad, S., Takas, M.E. Synthesis and characterization of porous clay heterostructure intercalated with CuO nanoparticles as a visible light-driven photocatalyst. J IRAN CHEM SOC 16, 45–55 (2019). https://doi.org/10.1007/s13738-018-1479-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-018-1479-8