
Abstract
In this work, a new and simple method is proposed to estimate the melting points of hazardous organic peroxide compounds including hydroperoxides, dialkyl peroxides, primary and secondary ozonides, peroxyacids, diacyl peroxides and alkyl peroxyesters compounds. This method can be applied for any peroxide compound with general formula CxHyOz to predict its melting point on the basis of elemental composition and specific structural moieties as additive and non-additive functions. It was applied for 104 different peroxide compounds including complex molecular structures. The predicted results give more reliable results in comparison to two of the best available methods. The average and maximum percent deviations of the new method are 6.8 and 21.1, respectively, which are lower than corresponding predicted values of Joback–Reid (24.1 and 101.9) and Jain–Yalkowsky (25.4 and 211.1).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The debate about new energetic compounds with suitable thermodynamic properties, performance and sensitivity is of utmost importance to scientists. Moreover, synthesis and measuring of properties of energetic compounds are dangerous, expensive, time consuming or sometimes impossible [1].
Prediction of melting point of energetic materials can be done by quantum mechanical calculations [2–5]. This approach has been considered difficult through simulation because of the presence of the free energy barrier for the formation of a liquid–solid interface. Methods based on quantitative structure property (QSPR) and some empirical models have been developed for organic molecules including a number of drugs and/or homologous series [6–13].
For energetic materials, computer codes and empirical methods can help to improve systematic and scientific formulation of appropriate futuristic target molecules having enhanced performance as well as good thermal stability, impact and friction sensitivity. Empirical methods can be used to confirm the computer output through desk calculations of performance and physicochemical properties of energetic compounds. Due to the expenditure connected with the development and synthesis of a new energetic material [14–16], development of reliable methods is essential for prediction of desired properties of energetic materials, e.g., enthalpy of fusion [17–22]. Moreover, the knowledge of physical properties and performance of energetic materials can be useful for new and complex synthesis.
Prediction of melting point is important for using in chemical identification, purification and calculation of the other physicochemical properties such as vapor pressure and aqueous solubility [12]. Group contribution methods can be used to estimate melting points of different classes of organic compounds [23], which are based on the sum of contributions of small groups of atoms constituting the molecule. For example, some group contribution methods are Lydersen [24], Ambrose [25], Klincewicz and Reid [26], Joback and Reid [27], Lyman et al. [28], Horvath [29], Prickett et al. [30], Constantinou et al. [31–33], Marrero-Morej´on and Pardillo-Fontdevilla [34], Marrero and Gani [35]. However, in contrast to the other physicochemical properties, prediction of melting points was not very well estimated by the group contribution methods [6, 24, 36, 37]. In addition, there is no reliable method for predicting melting points of hazardous materials containing peroxide bonds.
The purpose of this work is to introduce a new simple model for prediction of melting points of organic peroxides, which can be classified according to their molecular structures as hydroperoxides, dialkyl peroxides, α-oxygen substituted alkyl hydroperoxides and dialkyl peroxides, primary and secondary ozonides, peroxyacids, diacyl peroxides (acyl and organosulfonyl peroxides), and alkyl peroxyesters (peroxycarboxylates, peroxysulfonates and peroxyphosphates) [38]. The predicted results of the new model were also compared with two of the best available methods, e.g., Joback–Reid (JR) [27] and Jain–Yalkowsky (JY) [10] methods.
Results and discussion
The study of various organic compounds containing peroxide bonds has shown that the elemental composition has an important contribution in prediction of their melting point. The study of compounds containing –O–O– groups has indicated that it is possible to express the melting points of these compounds as core and correcting functions [37]:
where \( T_{\text{m,peroxide}} ,\,T_{\text{core}} \) and \( T_{\text{correcting}} \) are melting points of peroxide compound, core and correcting functions, respectively. The parameter \( T_{\text{core}} \) is due to the contribution of elemental composition. The factor \( T_{\text{correcting}} \) is a correcting function that can be specified on the basis of molecular structure of desired peroxide molecule. The presence of some specific polar groups such as –OH or more than one peroxy acid groups without any functional groups may enhance intermolecular interactions. In contrast, the presence of some specific molecular moieties under certain conditions can decrease molecular attractions. Table 1 contains experimental data of melting points for 104 peroxide organic compounds, which have been used to optimize Eq. (1) with respect to different functional groups and molecular fragments. Multiple linear regression method was used to obtain the relative contributions of elemental composition in \( T_{\text{core}} \) and molecular moieties in \( T_{\text{non - add}} \) [37]. However, a general correlation for any peroxide organic compound can be introduced as:
where \( n_{\text{C}} ,\,n_{\text{H}} \) and \( n_{\text{O}} \) are the number of carbon, hydrogen and oxygen atoms, respectively; \( T_{\text{m,peoroxide}}^{ + } \) and \( T_{\text{m,peoroxide}}^{ - } \) are the positive and negative contributions of structural parameters in \( T_{\text{correcting}} \), respectively. As seen in Eq. (2), two parameters \( T_{\text{m,peoroxide}}^{ + } \) and \( T_{\text{m,peoroxide}}^{ - } \) can correct the values obtained on the basis of the contribution of elemental composition for the existence of several molecular fragments.
Two parameters \( T_{\text{m,peoroxide}}^{ + } \) and \( T_{\text{m,peoroxide}}^{ - } \)
For the presence of several molecular moieties, the values of \( T_{\text{m,peoroxide}}^{ + } \) and \( T_{\text{m,peoroxide}}^{ - } \) can be specified.
Prediction of \( T_{\text{m,peoroxide}}^{ + } \)
The existence of hydrogen bonding polar –OH or more than one peroxy acid groups without any functional groups can lead to much more efficient packing and the attractive forces confining the respective species in the crystal lattice. This situation was also confirmed in previous studies for different classes of energetic compounds [39]. For different aromatic and non-aromatic organic compounds containing these molecular moieties, there is a reinforced intermolecular hydrogen bond. Since dipole moment is an important factor for controlling the melting point, the sum of local dipole moments has a more pronounced effect than net dipole moment on melting point for some specific molecular fragments. The values of \( T_{\text{m,peoroxide}}^{ + } \) are 2.0 and 0.5 for the existence of more than one peroxy acid group without any functional groups and –OH, respectively.
Prediction of \( T_{\text{m,peoroxide}}^{ - } \)
For some organic molecules including –(CO)OO– and –O–C(O)–OO–(CO)–O– groups, the values of \( T_{\text{core}} \) are higher than experimental data. The presence of these groups may reduce the packing efficiency of molecules in the crystals, which can decrease the interaction between local dipole moments of neighboring polar groups. For the presence of only one –(CO)OO– or –O–C(O)–OO–(CO)–O– in form R1–(CO)OO–R1 or R1–O–C(O)–OO–(CO)–O–R1, the value of \( T_{\text{m,peoroxide}}^{ - } \) is 1.0 where R1 in both side of organic molecule should be the same. The value of \( T_{\text{m,peoroxide}}^{ - } \) also equals 1.0 for R–C(O)OOH where the number of carbon atoms in R should contain less than five carbon atoms.
Statistical parameters and reliability of Eq. (2)
Table 2 shows statistical parameters of Eq. (2) corresponding to five variables of \( n_{\text{C}} ,\,\,n_{\text{H}} ,\,\,n_{\text{O}} ,\,\,T_{\text{m,peoroxide}}^{ + } \) and \( T_{\text{m,peoroxide}}^{ - } \). It allows comparing the relative weight of the variables in the model. As indicated in Table 2, each of \( n_{\text{C}} ,\,\,n_{\text{H}} ,\,\,n_{\text{O}} ,\,\,T_{\text{m,peoroxide}}^{ + } \) and \( T_{\text{m,peoroxide}}^{\text{ - }} \) has a highly significant impact as evidenced by their extremely small p values and standard errors. Standard error is a measure of the precision of evaluation of a coefficient in which precision can be measured by standard deviation over repeated measurements. Meanwhile, the p value measures the probability that a parameter estimated from experimental data should be as large as it is. For p value <0.05, the observed effect is not due to random variations and the effect is significant. Eq. (2) is a good correlation because its R-squared value or the coefficient of determination is 0.970 [40].
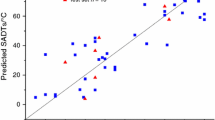
As indicated in Table 1, the predicted results of the new method for various peroxide compounds were also compared with JR [27] and JY [10] methods. The average absolute deviations of the new, JR [27] and JY [10] methods are 6.76, 24.14 and 25.42, respectively, which confirm high reliability of the new method with respect to both JR [27] and JY [10] methods. A visual comparison of the predicted results of the new, JR [27] and JY [10] methods with the experimental values is shown in Fig. 1. The predicted results of the new model show that it can be easily applied to molecular structures of different classes of peroxide compounds. Comparison of deviations of different classes of organic peroxides is given in Table 3. As seen, the best predictive result is related to peroxyacids, i.e., 5.9. For the other classes of organic peroxides, the proposed method can also provide good predictions because the difference between maximum and minimum of deviations is 1.8, which can be ignored.

Calculated melting points versus experimental data for different 104 peroxide compounds given in Table 1. The solid lines represent exact agreement between predictions and experiment. Filled circle, hollow circle and filled triangle denote the calculated values of the new, JR [27] and JY [10] methods, respectively
Conclusions
A novel method has been developed for the simple and reliable prediction of melting points of hazardous peroxide compounds including hydroperoxides, dialkyl peroxides, primary and secondary ozonides, peroxyacids, diacyl peroxides and alkyl peroxyesters compounds. The methodology presented here is based on a melting core temperature as well as \( T_{\text{m,peoroxide}}^{ + } \) and \( T_{\text{m,peoroxide}}^{ - } \) correction terms. As shown in Table 1, the new method gives more reliable predictions as compared to JR [27] and JY [10] methods, which may be taken as appropriate validation of the new method.
Since prediction of melting point of peroxide material is readily calculated in the new method by a desk calculator, the results of this study are appealing to chemists. The new model gives the simplest and easiest pathway for calculation of melting point of peroxide compounds. This reliable method confirms that the accuracy is not necessarily enhanced by greater complexity.
Abbreviations
- \( T_{\text{m,peroxide}} \) :
-
Melting points of peroxide compound
- \( T_{\text{core}} \) :
-
Additive core function
- \( T_{\text{correcting}} \) :
-
Non-additive molecular fragments
- \( T_{\text{m,peoroxide}}^{ + } \) :
-
Positive contribution of structural parameters in \( T_{\text{correcting}} \)
- \( T_{\text{m,peoroxide}}^{ - } \) :
-
Negative contribution of structural parameters in \( T_{\text{correcting}} \)
- \( n_{\text{C}} \) :
-
The number of carbon atoms
- \( n_{\text{H}} \) :
-
The number of hydrogen atoms
- \( n_{\text{O}} \) :
-
The number of oxygen atoms
References
J.P. Agrawal, High Energy Materials: Propellants, Explosives and Pyrotechnics (Wiley, USA, 2010)
P.M. Agrawal, B.M. Rice, D.L. Thompson, J. Chem. Phys. 119, 9617 (2003)
S. Alavi, D.L. Thompson, J. Phys. Chem. B 109, 18127 (2005)
P.M. Agrawal, B.M. Rice, L. Zheng, G.F. Velardez, D.L. Thompson, J. Phys. Chem. B 110, 5721 (2006)
A. Siavosh-Haghighi, D.L. Thompson, J. Phys. Chem. C 111, 7980 (2007)
P. Simamora, S.H. Yalkowsky, Ind. Eng. Chem. Res. 33, 1405 (1994)
D. Mackay, R.S. Boethling, Handbook of Property Estimation Methods for Chemicals: Environmental Health Sciences (CRC, USA, 2000)
J.S. Chickos, G. Nichols, J. Chem. Eng. Data 46, 562 (2001)
A. Jain, G. Yang, S.H. Yalkowsky, Ind. Eng. Chem. Res. 43, 7618 (2004)
A. Jain, S.H. Yalkowsky, J. Pharm. Sci. 95, 2562 (2006)
A. Jain, S.H. Yalkowsky, Ind. Eng. Chem. Res. 46, 2589 (2007)
Q. Wang, P. MA, S. NENG, Chin. J. Chem. Eng. 17, 468 (2009)
D.C. Evans, S.H. Yalkowsky, Fluid Phase Equilib. 303, 10 (2011)
M.H. Keshavarz, M. Oftadeh, High Temp. High Press. 35, 499 (2004)
M.H. Keshavarz, J. Hazard. Mater. 136, 145 (2006)
M.H. Keshavarz, J. Hazard. Mater. 136, 425 (2006)
M. Keshavarz, Indian J. Eng. Mater. Sci. 14, 386 (2007)
M.H. Keshavarz, J. Hazard. Mater. 150, 387 (2008)
M.H. Keshavarz, H.R. Pouretedal, Fluid Phase Equilib. 298, 24 (2010)
A. Semnani, M.H. Keshavarz, J. Hazard. Mater. 178, 264 (2010)
M.H. Keshavarz, Propellants Explos. Pyrotech. 36, 42 (2011)
Y. Mosaei Oskoei, M.H. Keshavarz, Fluid Phase Equilib. 326, 1 (2012)
B.E. Poling, J.M. Prausnitz, O.C. John Paul, R.C. Reid, The Properties of Gases and Liquids (McGraw-Hill, New York, 2001)
A. Lydersen, R.A. Greenkorn, O.A. Hougen, Estimation of Critical Properties of Organic Compounds by the Method of Group Contributions (University of Wisconsin, USA, 1955)
D. Ambrose, Correlation and Estimation of Vapour–Liquid Critical Properties (National Physical Library, UK, 1978)
K. Klincewicz, R. Reid, AIChE J. 30, 137 (1984)
K.G. Joback, R.C. Reid, Chem. Eng. Commun. 57, 233 (1987)
W.J. Lyman, W.F. Reehl, D.H. Rosenblatt, D.H. Rosenblatt, Handbook of Chemical Property Estimation Methods: Environmental Behavior of Organic Compounds (McGraw-Hill, New York, 1982)
G. Tari, F. Horváth, J. Rumpler Tectonophys. 208, 203 (1992)
L. Constantinou, S.E. Prickett, M.L. Mavrovouniotis, Ind. Eng. Chem. Res. 32, 1734 (1993)
S.E. Prickett, L. Constantinou, M.L. Mavrovouniotis, Mol. Simul. 11, 205 (1993)
L. Constantinou, R. Gani, AIChE J. 40, 1697 (1994)
L. Constantinou, S.E. Prickett, M.L. Mavrovouniotis, Ind. Eng. Chem. Res. 33, 395 (1994)
J. Marrero-Morejón, E. Pardillo-Fontdevila, AIChE J. 45, 615 (1999)
J. Marrero, R. Gani, Fluid Phase Equilib. 183, 183 (2001)
J.F. Krzyzaniak, P.B. Myrdal, P. Simamora, S.H. Yalkowsky, Ind. Eng. Chem. Res. 34, 2530 (1995)
R.F. Alamdari, M.H. Keshavarz, Fluid Phase Equilib. 292, 1 (2010)
E. Kirk-Othmer, Encyclopedia of Chemical Technology (Wiley-Interscience, USA, 2004)
M.H. Keshavarz, J. Hazard. Mater. 171, 786 (2009)
W.J. Palm, Introduction to MATLAB 7 for Engineers (McGraw-Hill, New York, 2005)
Acknowledgments
We would like to thank the research committee of Kashan University for supporting this project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khozani, M.H., Keshavarz, M.H., Nazari, B. et al. Simple approach for prediction of melting points of organic molecules containing hazardous peroxide bonds. J IRAN CHEM SOC 12, 587–598 (2015). https://doi.org/10.1007/s13738-014-0516-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-014-0516-5