
Abstract
A simple and efficient liquid-phase microextraction technique was developed using ultrasound-assisted emulsification solidified floating organic drop microextraction combined with flame atomic absorption spectrometry, for the extraction and determination of trace amounts of iron and copper in real samples. 2-Mercaptopyridine n-oxide was used as chelating agent and 1-dodecanol was selected as extraction solvent. The factors influencing the complex formation and extraction were optimized. Under optimum conditions, an enrichment factor of ~13 was obtained for both iron and copper from only 6.7 mL of aqueous phase. The analytical curves were linear between 40–800 and 20–1,200 μg L−1 for iron and copper respectively. Based on three SD of the blank, the detection limits were 8.6 and 4.1 μg L−1 for iron and copper respectively. The relative SDs for ten replicate measurements of 500 μg L−1 of metal ions were 2.9 and 1.2 for iron and copper respectively. The proposed method was successfully applied for determination of iron and copper in environmental waters and some food samples including chess, rice, honey and powdered milk. Finally, method validation was made using rock certified reference material. A student’s t test indicated that there was no significant difference between experimental results and certified values.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Trace metals play an important role in human metabolism and either excess or deficiency of them in the living organism can lead to biological disorder [1, 2]. Copper is both vital and toxic for many biological systems. According to national surveys, the average dietary intake of copper in the US is approximately 1.0–1.1 mg/day for adult women and 1.2–1.6 mg/day for adult men. Diary ingestion of copper is indispensable for good health. However, high amounts of copper can be harmful, causing irritation of nose and throat, nausea, vomiting, and diarrhea. Very high doses of copper can cause damage to liver and kidneys [3–5]. Copper is considered an essential trace element for plants and animals. Some compounds are toxic by ingestion or inhalation. The United Nations Food and Agriculture Organization recommended maximum level for irrigation waters is 200 μg L−1. Under the lead–copper rule, the U.S. EPA drinking water 90th percentile action level is 1.3 mg L−1 [6].
Iron is an essential and useful element for organism and an important part of tissue and blood in animal and human being. Iron is mainly distributed in hemachrome, which occupied 60–70 % of total iron in a body. A lack of iron can lead to iron deficiency anemia. Iron is stored in the body and large amounts can be toxic. High doses of iron can cause nausea, vomiting, stomach pain and constipation. National survey data shows that in Britain, on average, adult men consume 13.2 mg iron/day and women 10.0 mg iron/day from food sources [7]. Elevated iron levels in water can cause stains in plumbing, laundry, and cooking utensils, and can impart objectionable tastes and colors to foods. The United Nations Food and Agriculture Organization recommended level for irrigation waters is 5 mg L−1. According to the United State Environmental Protection Agency (U.S. EPA) secondary drinking water standard, Maximum Contaminant Level (MCL) of iron is 0.3 mg L−1 [6].
The direct determination of trace elements by modern atomic spectroscopic methods, such as flame atomic absorption spectrometry (FAAS) and inductively coupled plasma atomic emission spectrometry (ICP-AES) is often difficult because of insufficient sensitivity, selectivity and the interfering effects sources from the matrix of the real samples. Therefore, it is very important to develop sensitive methods for quantitative determination of trace copper and iron in various matrices. For this reason, the preliminary separation and preconcentration of trace elements from the matrix are often required. Many preconcentration procedures for copper and iron determination have been developed and they involve different analytical techniques and several materials. Among the methods of copper preconcentration reported are liquid–liquid extraction(LLE) using dithiocarbamate [8] and trioctylmethylammonium chloride [9] as complexing reagents, coprecipitation with magnesium hydroxide as collector [10] and also solid phase extraction (SPE) that use sorbents such as polyurethane foam loaded with diethyldithiocarbamate [11], activated carbon [12], Amberlite XAD resins [13, 14] and naphthalene [15]. Several preconcentration methods such as co-precipitation with magnesium hydroxide as collector [16], LLE using 4-acetyl-5-methyl-1-phenyl-1H-pyrazole-3-carboxylic acid as complexing reagents [17] and SPE that use sorbents such as silica gel [18] and Octadecyl-bonded silica membrane disk [19], have been developed for the separation and preconcentration of iron from environmental matrices.
However, the use of classical extraction methods for these purposes are usually time-consuming, labor-intensive and requires large amounts of high purity solvents for extraction. In recent years there has been a growing interest in the development of miniaturized preconcentration methods based on LLE or SPE [20–23] an approach that allows high preconcentration factors to be obtained as defined previously. One of these techniques is the so called liquid phase microextraction by solidification of a floating organic droplet (LPME-SFO) [24–26]. This technique has been successfully applied using undecanol and dodecanol to concentrate substances like organochlorine [27], and organophosphorus pesticides [28], pyrazoline derivatives [29] and some metals like lead [30], cadmium [26], cobalt and nickel [31]. It is well known that ultrasound is a powerful energy for the acceleration of various steps in analytical procedures, such as homogenizing and emulsion forming [32]. This type of energy greatly helps in the processes of separation and extraction because it facilitates accelerates the mass-transfer process between two immiscible phases [33]. This leads to an increment in the extraction efficiency of the procedure in a minimum time [34]. As a result, the application of an ultrasound radiation to a miniaturized approach such as solidification of a floating organic droplet (SFODME) provides the advantages of both methods [25]. This new technique is called ultrasound-assisted emulsification solidified floating organic drop micro extraction (USAE-SFODME). This method offers advantages of such as simplicity, low cost; rapidity, high enrichment factor, and low consumption of the extraction solvent. Recently, determination of trace amounts of zinc and cadmium in water samples were successfully performed with this method [35, 36].
The aim of this study was the development of a green analytical method for extraction and determination of iron and copper with use of 2-mercaptopyridine-n oxide as a selective complexing reagent. The combination of ultrasound energy with solidified floating organic drop procedure was employed for extraction of copper and iron with minimum consumption of reagents. Results obtained in the analysis of samples of water, food and rock are reported to demonstrate the effectiveness of the method.
Experimental
Analytical instrumentation
Determination of iron, copper and other cations were performed on a Shimadzu AA-670 atomic absorption spectrometer (Kyoto, Japan) under the recommended condition (wavelengths 248.3 and 324.8 nm, and bandwidths 0.2 and 0.5 nm, for iron and copper, respectively). All pH measurements were made using a Metrohm E-691 digital pH meter with a combined glass electrode. A model Labofuge 400 (Germany) centrifuge was used to accelerate phase separation. A model Parasonic 7500S, 28 kHz, 100 W ultrasonic bath with temperature control was used to assist the emulsification process of the microextraction technique.
Reagents and standard solutions
Nitric acid, hydrochloric acid, hydrogen peroxide, sulfuric acid, formic acid, perchloric acid, hydrofluoric acid and 1-dodecanol from Merck were used as received. The stock solution of 1,000 mg L−1 of iron and copper were prepared by dissolving of Fe(NO3)3·9H2O and Cu(NO3)2·3H2O (Merck) in 2 mL concentrated nitric acid (65 % Merck). The solution reached to the mark in 1,000 mL volumetric flask with deionized water. A 1.0 g L−1 solution of 2-mercaptopyridine n-oxide sodium salt (Alfa Aesar, Germany) (Scheme 1) in water was prepared. Other metal salts were analytical grade and purchased from Merck. Working solutions were prepared by appropriate dilution of the stock solution.

The structure of 2-mercaptopyridine n-oxide sodium salt (pyrithion)
Extraction procedure
In a 10 mL centrifuge tube, 5.0 mL of 500 μg L−1 Fe(III) and Cu(II) solution or real samples, 1.0 ml of formate buffer 0.1 M (concentration in solution is 0.015 M), and 0.7 mL of 2-mercaptopyridine n-oxide solution in water (1.0 g L−1) were mixed. Then, 100 μL of 1-dodecanol was added using a 100 μL syringe. The conical tube was sonicated for 20 min at 40 ± 3 °C to ensure complete extraction. The mixture was then centrifuged for 5 min at 3,500 rpm. After this process, fine droplets of 1-dodecanol coalesced and the organic solvent collected at the upper surface of sample solution. The conical test tube was transferred into an ice bath and the organic solvent was solidified after 3 min and adhered on inner surface of the tube. The aqueous phase was easily decanted into another vial (without using a spatula) and the remaining solid solvent melted immediately. Next, the extract was diluted to 500 μL with ethanol and manually injected into the flame atomic absorption spectrometer [35, 36]. Figure 1 shows a scheme of the USAE-SFODME procedure.

Schematic diagram for the proposed USAE-SFODME procedure
Preparation of natural waters
The river water from Sepid Rood (Lahijan, IRAN), mineral water (Hayat, IRAN) and drinking water (Sanandaj, IRAN) were acidified to pH < 2.0 with concentrated HNO3, immediately filtered (for river water) and stored in precleaned polyethylene bottles. In order to determine the total iron and copper, a 50.0 mL aliquot of each sample was oxidized by addition of 5.0 mL concentrated HNO3 and 1.0 mL concentrated H2O2 (30 %). The beaker was covered with watch glass and heated at 100 °C for 30 min to complete the oxidation [37]. Then 5 mL of this solution was tested for determination of iron and copper under the general procedure.
Preparation of food samples
5.00 g of food sample was weighed and was put in a 200 mL beaker and then 10.0 ml of concentrated HNO3 was added. The sample was then heated up to 95 °C for about 1 h until a yellow solution appeared. The sample was cooled and then 5.0 mL of concentrated sulfuric acid was added and then heated up to 140 °C until the first sign of charring was appeared. The food sample was then cooled again, 5.0 mL concentrated nitric acid was added and the sample was heated up to 180 °C. Further 1.0 mL of nitric acid was added until the digested sample was appeared clear or a pale-straw color was observed. The sample was then cooled, 1.0 mL H2O2 (500 g L−1) was added and heated up to 200 °C. This procedure was repeated until brown fumes ceased to be appeared. After the sample was cooled, 10.0 mL of water and 0.5 mL of concentrated nitric acid was added, and the sample was heated up to 200 °C until white fumes were evolved. To the sample solution 10.0 mL of water, followed by 1.0 mL H2O2 (500 g L−1) was added and heated again up to 240 °C until white fumes were evolved. Finally, the digested sample was cooled and quantitatively transferred to a 100.0 mL volumetric flask for analysis [38]. For the analysis of concentrated samples another dilution was done. After adjusting the pH, analysis was done with 5 mL sample as previously mentioned.
Preparation of rock certified reference material
The rock sample analyzed according to Refs. [39, 40] with a little modification: 0.1 g of powdered rock was weighed in a 50 mL Teflon beaker and 4 mL HNO3 (65 %), 3 ml HClO4 (70 %) and 5 mL HF (40 %) were added. These were mixed well and this mixture was kept for more than 30 min, and then the beaker was covered and heated at ~160 °C for 1 day. Then the mixture was recovered and evaporated to dryness at ~140 °C for 2–3 days. The residue was dissolved with 10 mL (1 + 1) HCl by heating and dilution to 50 mL for analysis. After another dilution (1,000 and 500 times for Fe and Cu respectively) and adjusting the pH, analysis was done as previously mentioned.
Results and discussion
Selection and effect of volume of the extraction solvent
The organic solvent used as the extracting solvent in this method has to satisfy several criteria: (1) it must be immiscible with water and have low volatility in order to be stable during the extraction period and have good extraction efficiency, (2) it should have a lower density than water, (3) it should have a melting point near room temperature (in the range of 10–30 °C) [26, 41]. Therefore 1-dodecanol (density 0.8201–0.8309 g mL−1; melting point 24 °C) was used as extraction solvent.
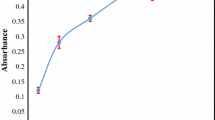
During SFODME based on USAE process, extracting solvent volume was an essential factor which could influence the occurrence of the emulsion state and also determine enrichment performance. To evaluate the effect of the extraction solvent volume, different volumes of 1-dodecanol were added to 7.0 mL aqueous phase (5.0 ml of sample solution containing 500 μg L−1 of Fe(III) and 500 μg L−1 Cu(II) ions and 2.0 mL ligand 1.0 g L−1) in the range of 20–160 μL. The results are shown in Fig. 2. As can be seen, absorbance increased with the increase of 1-dodecanol volume in the range of 20–80 μL, and then remained constant when the volume was continuously increased. Therefore, in the subsequent studies, 100 μL was selected as the optimum volume of the extraction solvent. After the preconcentration procedure, the obtained volume of 1-dodecanol was 100 ± 4 μL.

Effect of volume of extraction solvent on absorbance of iron and copper. Condition: water sample volume, 5.0 mL; 2.0 mL 2-mercaptopyridine n-oxide 1.0 g L−1; extraction solvent (1-dodecanol); concentration of Fe(III) and Cu(II), 500 (μg L−1); extraction time, 20 min; extraction temperature 40 °C; N = 3
Influence of pH
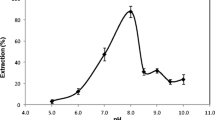
The separation of metal ions by USAE-SFODME involves prior formation of a complex with sufficient hydrophobicity that allows it to be extracted into the small volume of the floated phase, where the desired preconcentration is obtained. So, the effect of pH on the complex formation and extraction of Fe3+ and Cu2+ from 7.0 mL of aqueous phase into organic phase (100 μL 1-dodecanol) was studied in the range of 1.0–9.0. The pH values were adjusted either by nitric acid or sodium hydroxide solution. The experimental results illustrated in Fig. 3 show that the maximum absorbance was obtained at pH 3.0 for iron and copper. The decrease in extraction of iron and copper ions at higher pHs may be due to competition of hydroxyl ion with pyrithione for reaction with analytes, and in lower pHs is due to protonation of ligand at these pHs. Therefore a pH 3.0 was chosen for subsequence experiments and the pH adjustment was carried out by formate buffer solution.

Effect of pH on absorbance of iron and copper by USAE-SFODME. Condition: water sample volume, 5.0 mL; 2.0 mL 2-mercaptopyridine n-oxide 1.0 g L−1; extraction solvent (1-dodecanol) 100 μL; concentration of Fe(III) and Cu(II), 500 (μg L−1); extraction time, 20 min; extraction temperature 40 °C; N = 3
Effect of amount of 2-mercaptopyridine n-oxide
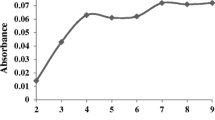
The effect of amount of 2-mercaptopyridine n-oxide on the extraction efficiency of iron and copper from 6.0 mL of aqueous phase (5.0 ml of sample solution containing 500 μg L−1 of Fe3+ and 500 μg L−1 of Cu2+ ions and 1.0 mL 0.1 M format buffer) into organic phase (100 μL 1-dodecanol) was studied using various amounts of ligand (1.0 g L−1) ranging from 0.0 to 1.8 mg. The absorbance was stable when the ligand amount was higher than 0.6 mg, indicating complete complexation (Fig. 4). Therefore, 0.7 mg 2-mercaptopyridine n-oxide (equal 0.7 mL of 1.0 g L−1 solution) was chosen as the optimum amount for the iron and copper extraction.

Effect of amount of 2-mercaptopyridine n-oxide on absorbance of iron and copper. Condition: water sample volume, 5.0 mL; 1.0 mL formate buffer 0.1 M; extraction solvent (1-dodecanol) 100 μL; concentration of Fe(III) and Cu(II), 500 (μg L−1); extraction time, 20 min; extraction temperature 40 °C; N = 3
Effect of sonication time
To increase the precision and sensitivity of the USAE-SFODME method, it is necessary to select an exposure time that guarantees the equilibrium between aqueous and organic phases has been achieved. Thus the effect of sonication time on extraction efficiency of iron and copper from 6.7 mL of aqueous phase (5.0 mL of sample solution containing 500 μg L−1 of Fe3+ and 500 μg L−1 of Cu2+ ions, 1.0 mL 0.1 M format buffer and 0.7 ml 2-mercaptopyridine n-oxide solution) into organic phase (100 μL 1-dodecanol) was examined in the range of 0–30 min with a constant ultrasonic frequency. It was seen that maximum FAAS signal for both iron and copper reached at ~20 min (Fig. 5), which was chosen as the sonication time for further experiments. Hence, 20 min was chosen for the further experiments as the sonication time.

Effect of sonication time on absorbance of iron and copper by USAE-SFODME. Condition: water sample volume, 5.0 mL; 1.0 mL formate buffer 0.1 M; 0.7 mL 2-mercaptopyridine n-oxide 1.0 g L−1; extraction solvent (1-dodecanol) 100 μL; concentration of Fe(III) and Cu(II), 500(μg L−1); extraction temperature 40 °C; N = 3
Extraction temperature
Temperature affects organic solvent solubility in water as well as the emulsification phenomenon. Thus, it also affects the mass-transfer process and the extraction efficiency [41]. To determine the influence of the extraction temperature, 6.7 mL of aqueous phase was extracted with organic phase (100 μL 1-dodecanol) at different temperatures ranging from 25 to 50 °C. The results revealed that the absorbance was increased by increasing the extraction temperature up to 35 °C. At temperature of lower than 35 °C, it was difficult to get a homogeneous emulsion resulting in a prompt phase separation. Therefore, the mass transfer process was limited to a short time, leading to poor extraction efficiency, and consequently low absorbance [41]. In the temperature range of 35–50 °C, the emulsification was easily achieved and the obtained absorbance remained constant at this temperature range. Hence, 40 °C was chosen for further studies.
Effect of salt
For studying the influence of ionic strength on the efficiency of USAE-SFODME, various experiments were performed by adding varying NaCl amounts from 0 to 5 % (w/v) [41] while other experimental conditions were kept constant. Increasing the NaCl concentration had no significant effect on extraction factor, perhaps because of the two opposite effects of salt addition in USAE-SFODME of iron and copper: One involves increasing the volume of the floated phase, which decreases the enrichment factor, and the other is the salting-out effect that increases the enrichment factor [42, 43]. Therefore, the enrichment factor is nearly constant by increasing the amount of sodium chloride, and the extraction experiments were carried out without additional salt.
Effect of diverse ions
The effects of common coexisting ions on the recovery of iron and copper were also studied. In these experiments, 5.0 mL of solutions containing 500 μg L−1 of metal ions and various amounts of diverse ions were treated according to the recommended procedure. A given species was considered to interfere if it resulted in a ±5 % variation of the absorbance signal. The results were given in Table 1. As can be seen from Table 1, majority of the investigated ions have no significant influence on extraction of Fe(III) and Cu(II) under the selected conditions. This may be due to formation of more stable complexes of Fe(III) and Cu(II) with pyrithione than the other metal ions studied. Lofts showed that the order of stability constants and hence, reactivity of the pyrithione toward metal ions follows the trend of Fe(III) > Cu(II) > Pb(II) > Zn(II) > Ni(II) > Co(II) > Cd(II) > Mn(II) > Ca(II) [44]. Thus it is not surprising that other cations can’t significantly interfere in the extraction of Fe(III) and Cu(II) from aqueous solution.
Analytical figures of merit
Using the optimum conditions described above, the analytical characteristics of the proposed method, including linear range, limit of detection, relative SD, correlation coefficient (R 2), and enrichment factor, obtained by processing standard solutions of Fe(III) and Cu(II), are summarized in Table 2. The analytical curves were linear in the range of 40–800 and 20–1,200 μg L−1 with a correlation coefficient (R 2) of 0.9974 and 0.9994 for iron and copper respectively. The regression equations were A = 0.176 C + 0.0008 for iron and A = 0.344 C + 0.0002 for copper, where A is absorbance and C is the concentration of Fe3+ and Cu2+ in mg L−1. The limit of detections (LOD), calculated as the concentration of the absolute amount of analyte yielding a signal equivalent to three times the SD of the blank (n = 10, \( {\text{LOD}} = 3\sigma_{\text{blank}} /{\text{slope}} \)) in accordance to IUPAC recommendation, were 8.6 and 4.1 μg L−1 for the determination of Fe(III) and Cu(II) in 6.7 mL of initial aqueous phase (5.0 ml deionized water, 1.0 mL buffer and 0.7 mL of ligand).
Applications
The proposed method was successfully used for the determination of total iron and copper in several water and food samples. The results along with the recovery for the spiked samples were given in Tables 3, 4. As can be seen, added iron and copper are quantitatively recovered from water and food samples. The accuracy of the proposed method was evaluated by means of recovery experiments and analysis of certified reference material (JB-3, Basalt Geological survey of Japan). The results are shown in Table 5. These results indicate the validity of the proposed methodology for analysis of iron and copper in real samples.
Conclusion
Separation and determination of Fe(III) and Cu (II) by developed method were compared with the other reported preconcentration methods. The results are shown in Table 6. As can be seen, the proposed procedure shows good detection limit, wide linear dynamic range and lower sample consumption (5 mL), which are better in most cases and are comparable with reported methods in other cases. The newly developed USAE-SFODME method exhibited distinct advantages over the conventional extraction methods such as LLE and SPE. It is simple, rapid, inexpensive and environmentally friendly because low organic solvent consumption. In addition, the application of ultrasonic radiation is a key tool to improve the extraction efficiency of the microextraction procedure with minimal time. Moreover, the collection of the solidified phase from aqueous phase can be carried out easily and with a better precision than the other Ultrasound Assisted Emulsification Microextraction (USAEME) method [50]. The proposed method for simultaneous preconcentration and separation of iron and copper is selective and allow the determination trace amounts of these elements to be carried out by FAAS which is an available instrument in almost every laboratory. The method was successfully applied in water, food and rock samples.
References
B.C. Mondal, D. Das, A.K. Das, J. Trace Elem. Med Biol. 16, 145 (2002)
Ş. Tokalıoğlu, A. Livkebabcı, Microchim. Acta 164, 471 (2009)
Y. Yamini, A. Tamaddon, Talanta 49, 119 (1999)
N. Tokman, J. Hazard. Mater. 143, 87 (2007)
V.A. Lemos, E.S. Santos, E.M. Gama, Sep. Purif. Technol. 56, 212 (2007)
M.A.H. Franson, Standard method for examination of water and waste water. (American Publication Health Association. Washington, DC, 1995)
C. Xiong, Z. Jiang, B. Hu Anal, Chim. Acta 559, 113 (2006)
S. Sachsenberg, T. Klenke, W.E. Krumbein, E. Zeeck, J. Anal. Chem. 342, 163 (1992)
X.Y. Zhang, S. Keiichi, A. Satoh, K. Sawada, T. Suzuki, Anal. Sci. 13, 891 (1997)
J. Wu, E.A. Boyle, Anal. Chem. 69, 2464 (1997)
D. Atanasova, V. Stefanova, E. Russeva, Talanta 45, 857 (1998)
A. Uzawa, T. Narukawa, T. Okutani, Anal. Sci. 14, 395 (1998)
L. Elci, M. Soylak, M. Dogan, Fresenius J. Anal. Chem. 342, 175 (1992)
V.K. Jain, S.S. Sait, P. Shrivastav, Y.K. Agrawal, Talanta 45, 397 (1997)
M.A. Taher, J. Anal. Chem. 56, 149 (2001)
M. Grotti, F. Soggia, F. Ardini, R. Frache, J. Anal. At. Spectrom. 24, 522 (2009)
S. Sacmaci, S. Kartal, Anal. Chim. Acta 623, 46 (2008)
E. Pehlivan, D. Kara, Microchim. Acta 158, 137 (2007)
G. Khayatian, H. Ahmadzadeh Vosta Kolaie, F. Nasiri, B. Atashkar, S. Hassanpoor, J. Chin. Chem. Soc 57, 118 (2010)
F. Pena-Pereira, I. Lavilla, C. Bendicho, A review, Spectrochim. Acta B 64, 1 (2009)
C. Nerı′n, J. Salafranca, M. Aznar, R. Batlle, Anal. Bioanal. Chem 393, 809 (2009)
R.E. Rivas, I. Lopez-Garcia, M. Hernandez-Cordoba, Spectrochim. Acta B 64, 329 (2009)
Y. Liu, E.C. Zhao, W.T. Zhu, H.X. Gao, Z.Q. Zhou, J. Chromatogr. A 1216, 885 (2009)
H. Farahani, P. Norouzi, R. Dinarvand, M.R. Ganjali, J. Sep. Sci. 32, 314 (2009)
M. Mohamadi, A. Mostafavi, Talanta 81, 309 (2010)
M.R. Khalili-Zanjani, Y. Yamini, S. Shariati, J.A. Jonsson, Anal. Chim. Acta 585, 286 (2007)
H. Farahani, Y. Yamini, S. Shariati, M.R. Khalili-Zanjani, S. Mansour-Baghahi, Anal. Chim. Acta 626, 166 (2008)
M.R. Khalili-Zanjani, Y. Yamini, N. Yazdanfar, S. Shariati, Anal. Chim. Acta 606, 202 (2008)
H.R. Sobhi, Y. Yamini, A. Esrafili, M. Adib, J. Pharm. Biomed. Anal. 48, 1059 (2008)
S. Dadfarnia, A.M. Salmanzadeh, A.M.H. Shabani, Anal. Chim. Acta 623, 163 (2008)
M.S. Bidabadi, S. Dadfarnia, A.M.H. Shabani, J. Hazard. Mater. 166, 291 (2009)
M.E. Aydin, A. Tor, S. Ozcan, Anal. Chim. Acta 577, 232 (2006)
S. Ozcan, A. Tor, M.E. Aydin, Anal. Chim. Acta 647, 182 (2009)
M.D. Luque de Castro, F. Priego-Capote, Analytical applications of ultrasound (Elsevier, Amsterdam, 2006)
J.J. Ma, J.W. Zhang, X. Du, X. Lei, J.C. Li, Microchim. Acta 168, 153 (2010)
J. Zhang, Y. Wang, X. Du, X. Lei, J. Ma, J. Li, J. Braz. Chem. Soc. 22, 446 (2011)
A.S. Pereira, G. Ferreira, L. Caetano, M.A.U. Martines, P.M. Padilha, A. Santos, G.R. Castro, J. Hazard. Mater. 175, 399 (2010)
J.R. Dean, Method for environmental trace analysis (Wiley, England, 2003)
N. Imai, Anal. Sci. 6, 389 (1990)
H. Azizia, S. Chung, T. Tanaka, Y. Asahara, Precamb. Res. 185, 87 (2011)
Q. Chang, J. Zhang, X. Du, J. Ma, J. Li, Front. Environ. Sci. Eng. China 4, 187 (2010)
E.Z. Jahromi, A. Bidari, Y. Assadi, M.R.M. Hosseini, M.R. Jamali, Anal. Chim. Acta 585, 305 (2007)
A. Bidari, E.Z. Jahromi, Y. Assadi, M.R.M. Hosseini, Microchem. J. 87, 6 (2007)
S. Lofts, NERC Centre for Ecology and Hydrology, (CEH Project Number: C03634), (Unpublished), 2009
O. Yıldız, D. Citak, M. Tuzen, M. Soylak, Food Chem. Toxicol. 49, 458 (2011)
I. Narin, M. Soylak, Anal. Chim. Acta 493, 205 (2003)
D. Rekha, K. Suvardhan, K. Suresh Kumar, P. Reddyprasad, B. Jayaraj, P. Chiranjeevi, J. Serb. Chem. Soc. 72, 299 (2007)
A. Tabrizi, J. Hazard. Mater. 183, 688 (2010)
G. Khayatian, S. Pouzesh, J. Iran. Chem. Soc. 4, 490 (2007)
G. Khayatian, S. Hassanpoor, J. Chin. Chem. Soc. 59, 659 (2012)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Khayatian, G., Hassanpoor, S. Development of ultrasound-assisted emulsification solidified floating organic drop microextraction for determination of trace amounts of iron and copper in water, food and rock samples. J IRAN CHEM SOC 10, 113–121 (2013). https://doi.org/10.1007/s13738-012-0131-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-012-0131-2