
Abstract
Obesity is a multifactorial disorder that results in excessive accumulation of adipose tissue. Although obesity is caused by alterations in the energy consumption/expenditure balance, the factors promoting this disequilibrium are incompletely understood. The rapid development of new technologies and analysis strategies to decode the gut microbiota composition and metabolic pathways has opened a door into the complexity of the guest–host interactions between the gut microbiota and its human host in health and in disease. Pivotal studies have demonstrated that manipulation of the gut microbiota and its metabolic pathways can affect host’s adiposity and metabolism. These observations have paved the way for further assessment of the mechanisms underlying these changes. In this review, we summarize the current evidence for possible mechanisms underlying gut-microbiota-induced obesity. The review addresses some well-known effects of the gut microbiota on energy harvesting and changes in metabolic machinery, on metabolic and immune interactions, and on possible changes in brain function and behavior. Although there is limited understanding on the symbiotic relationship between us and our gut microbiome, and how disturbances of this relationship affects our health, there is compelling evidence for an important role of the gut microbiota in the development and perpetuation of obesity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The last few decades have seen a rapid increase in the worldwide prevalence of obesity [1]. This obesity epidemic has been associated with an increased incidence of metabolic syndrome, certain cancers, a reduction in the quality of life, and a dramatic increase in obesity-related health care costs [2, 3]. Even though many factors have been implicated in the increased prevalence of obesity, including the easy access to energy dense foods and a decrease in physical activity [2], mounting evidence supports an important role of alterations in the gut microbiome as a mediator of obesity [4].
The human gut, mainly the large bowel, harbors the greatest numbers of microbiota in the body when compared to other human-body niches such as the skin, vagina, mouth, and ears [5]. We carry two sets of genes: those encoding the human genome (about 23,000 genes) and those encoding our microbiota (about 3.3 million genes) [6, 7, 8•, 9]. Humans and microorganisms have long benefited from this symbiotic relationship, yet our understanding of the extent and meaning of this coexistence has been limited due to the lack of reliable and effective tools to study it [5]. Alterations in the gut microbiome have been shown to predispose its host to develop certain diseases, including obesity.
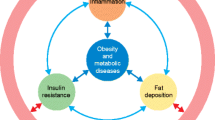
This paper is aimed to review current evidence of possible mechanisms associated with the gut microbiota in causing obesity. Following a brief overview of key findings supporting a relationship between changes in the gut microbiota and obesity, we will review the reported evidence for four mechanistic paths in gut-microbiota-mediated causes of obesity: changes in energy harvesting, changes in metabolic pathways, microbiota-induced inflammatory response, and possible changes in brain and behavior (Fig. 1).

Candidate mechanisms underlying gut microbiota induced obesity. Mechanistic pathways between the gut microbiota have been depicted with arrows. These include the following main pathways: (1) changes in energy harvesting, (2) changes in metabolic pathways, (3) the role of induced inflammatory responses, and (4) possible changes in brain and behavior. GPR G-protein receptor, LPS lipopolysaccharides, SCFA, short-chain fatty acids, TLR Toll-like receptors
Gut Microbiota and Obesity
The adult gut microbiota is dominated by two phyla, Firmicutes and Bacteroidetes, which classify about 90 % of all the bacterial species in the gut [5, 10]. Both in animal and human studies, obesity has been linked to a different composition of the gut microbiota. Many studies point towards a relative decrease in the abundance of Bacteroidetes together with a relative increase in the Firmicutes as a characteristic of the “obesogenic microbiota,” but the findings are far from consistent [11–15]. Obesity-related differences have also been identified at the species level including Clostridium innocuum, Eubacterium dolichum, and Catenibacterium mitsuokai, Lactobacillus reuteri, Lactobacillus sakei, Actinobacteria, and even members of Archae such as Methanobrevibacter smithii [10, 12, 14–18].
The lack of consistency in the reported results may in part be a reflection of the limitations of the current tools and study designs. One of the major open questions is whether the changes in intestinal microbiota precede the development of obesity or if they are a reflection of the obese phenotype. Due to the intricate interrelationships among diet, microbiota, immunity, and obesity development, it is a difficult question to answer. Changes in diet can cause obesity or weight loss and also concurrent changes in human gut microbiota composition, hence making it difficult to discern the relative role microbiota alone plays as the causative agent of changes in body weight. The ability to develop and preserve germ-free-mouse lines (gnotobiotic mice), which can be used as recipients of the gut microbiota from conventional mice (conventionalization) or from human donors (humanized mouse gut microbiota), has greatly accelerated progress in microbiome research. This germ-free model can be used to assess changes in recipients’ phenotypes, metabolism, and inflammation after fecal transplantation. Although the germ-free mouse has created unprecedented possibilities for mechanistic insights, there are significant limitations to its use including its intrinsic resistance to develop obesity, abnormal immune response, altered taste preferences, and brain and behavioral changes. In addition, despite successful initial colonization of germ-free mice with “lean or obese gut microbiota,” the composition of the gut microbiota is quickly changed by the diet of the recipient host [15, 19]. Regardless of these limitations, this model has been broadly used to study the role of the microbiome in obesity [15, 17, 20, 21].
Genetics and epigenetic events (including adverse early life events) contribute to an individual’s propensity to develop obesity, and these factors have also been shown to affect gut microbial composition [22–24]. These confounders can be partially controlled for in twin studies. For example, a study in twins discordant for body mass index (BMI) revealed a lower proportion of Bacteroidetes and a higher proportion of Actinobacteria in obese compared with lean individuals. However, there was no significant difference in the proportion of Firmicutes [14]. Using fecal transplantation of twins discordant for obesity in germ-free mice, Ridaura et al. [25••] were able to show transmission of the lean and obese phenotype. Cohabitation shows that the “lean microbiota” could successfully be transmitted into mice with “obese microbiota” and could partially prevent the development of obesity, suggesting that manipulation of gut microbiota can prevent the development of obesity. However, this was only the case while mice were consuming low-fat/high-fiber diets. Once the animals were exposed to high-fat/low-fiber diets, all mice experienced an increase in body mass and fat mass, and co-housing of the lean and obese mice failed to attenuate or block the development of obesity [25••]. These findings emphasize the importance of diet on obesity development, and the close interrelationship between diet and microbiota composition. Another well-studied factor influencing the development of obesity is the widespread use of antibiotics. Low doses of antibiotics have been used in farming to promote growth of livestock for several decades [26], and active or passive exposure to antibiotics is a likely factor contributing to the current obesity epidemic. Recent attention has been brought to the striking correlation between the geographical distribution of obesity and antibiotic use in the USA [27]. Other research has also shown that antibiotic exposure early in life is related to subsequent development of adiposity and obesity in human infants [28, 29] and in mice [30].
Candidate Mechanisms Underlying Gut Microbiota-Induced Obesity
Increased Energy Harvesting by the Gut Microbiota
Preclinical Studies
Obesity implies an imbalance between energy intake and expenditure, resulting in an excess of energy storage as adipose tissue. It has been proposed that the gut microbiota of obese individuals is more efficient at extracting energy from the diet than the microbiota of lean individuals. This hypothesis is supported by many studies showing an increase in body weight and fat in germ-free mice after transplanting gut microbiota derived from wild as well as from obese mice [25••, 11]. The weight gain is thought to be explained by several gut-bacteria-related mechanisms, including the microbial fermentation of indigestible dietary polysaccharides into absorbable monsaccharides, and the generation of short-chain fatty acids (SCFAs), which are converted to more complex lipids in the liver.
A number of studies have shown that, in both genetically obese mice (ob/ob) and diet-induced-obesity (DIO) mice, there is a relative reduction in the abundance of Bacteroidetes and a compensatory increase in the abundance of Firmicutes, compared to lean littermates [11, 13, 15]. Firmicutes are major producers of the SCFA butyrate. SCFAs are produced by bacterial fermentation of dietary carbohydrates and, to a lesser extent, of protein and peptides in the colon. The end products of this fermentation are SCFAs such as butyrate, propionate, and acetate together with gases (CO2, CH4, and H2) and heat [31]. Carbohydrates are fermented by saccharolytic bacteria primarily in the proximal colon-producing SCFAs, H2, and CO2. Fermentation of proteins and amino acids by proteolytic bacteria produce SCFAs, H2, CO2, CH4, phenols, and amines, which in turn influence the rate of cholesterol synthesis [32]. The main site of carbohydrate fermentation is the cecum and proximal colon where substrate and bacteria are available in larger proportions than in the distal colon [31, 33]. SCFAs are efficiently absorbed in the cecum and the colon with only 5–10 % being excreted in the feces [32]. Once absorbed, SCFAs are metabolized at three major sites in the body: (1) in the colonic epithelium that uses butyrate as a major substrate for energy; (2) in hepatocytes that use butyrate and propionate for gluconeogenesis and also take up most of the produced acetate that may be used for lipogenesis; and (3) in muscle cells that generate energy from acetate.
The theory of increased energy harvesting by the “obese microbiome” is supported by the finding of increased production of SCFAs in the cecum of obese mice and a decreased fecal energy content when compared to their lean counterparts. A seminal study for this hypothesis was done by Turnbaugh et al. [11], who compared fecal samples taken from the cecum of genetically obese mice (ob/ob) and wild-type littermates. The fecal samples showed an increase in the relative richness of Firmicutes in the ob/ob mice [11]. At 2 weeks, the ob/ob cecal fecal samples had an increased concentration of butyrate and acetate, and calorimetry revealed that ob/ob mice had significantly less energy remaining in their feces relative to their lean littermates. The metagenomic analysis in this study supported that the obesogenic fecal microbiome was rich in enzymes involved in the fermentation of undigestible polysaccharides. Moreover, there was an increase in methanogenic microorganisms (Archaea) in the ob/ob mice, which is known to improve efficiency in fermentation. Once the ob/ob fecal microbiota was transplanted by gavage into germ-free mice, there was an increase in Firmicutes in fecal samples as well as a small increase in body fat. These findings suggested the possibility that the capacity to develop obesity can be transmitted via fecal transplant, implicating the microbiome as an important factor in the development of obesity. However, several attempts to correlate the increased SCFAs production with a specific change at the species levels have demonstrated a lack of consistency among the studies [11, 16, 31, 34, 35].
There is growing evidence against a significant role played by increased microbial energy production/absorption as a major cause of obesity. First, there is evidence that the increased SCFAs production following fecal microbial transplant may only be transient. For example, while Murphy et al. [16] replicated the increase of SCFAs in the cecum shown by Turnbaugh et al. [11], SCFA levels in their study returned to normal by week 6. Furthermore, the SCFA increase was only observed in the ob/ob mice but not in the diet-induced obesity (DIO) mouse model [16]. The DIO mice showed an increase in relative proportion of Firmicutes in the cecum, but surprisingly, their fecal samples showed a sustained increase in fecal energy excretion. Neither fecal SCFAs nor fecal energy excretion correlated with relative proportions of Firmicutes or Bacteroidetes in this study [16]. Furthermore, high-fat and high-calorie diets have been associated with an increase in energy and SCFAs content in fecal samples in human and animal studies. It has been suggested that this compensatory mechanism aims to reduce weight gain when exposed to high-calorie diets.
Other possible mechanisms of gut-microbiota-related increase in the absorption of nutrients have been reported in rodent studies. High-fat diet has been associated with an increase in Erysipelotrichi, a class within the Firmicutes, and more specifically, Clostridium ramosum. In germ-free mice, C. ramosum has been shown to promote diet-induced obesity. In these studies, C. ramosium was also found to increase the expression of the glucose transporter 2 (Glut2) in jejunal mucosa and of the fatty acid translocase (CD36) in ileal mucosa, both of which could lead to increased absorption of carbohydrates and fat [17].
Methanogenic archaea increases the efficiency of bacterial fermentation by removing one of its end products, H2. Studies of gnotobiotic mice colonized with the methanogenic archaeon, M. smithii, and/or B. thetaiotaomicron revealed that co-colonization with these two microorganisms increases carbohydrate fermentation efficiency, a process that could lead to weight gain and obesity [10, 36, 37]. However, human studies with M. smithii have not confirmed the relationship of this organism with obesity [18, 36].
Human Studies
Schwiertz et al. [34] found significant differences in SCFAs concentration in fecal samples between normal lean, overweight, and obese volunteers. In these studies, fecal SCFA concentrations were more than 20 % higher in obese subjects than of lean volunteers. The proportion of propionate in the SCFAs was also higher in the feces of overweight and obese volunteers than in the feces of lean volunteers. Even though BMI was associated with propionate production, SCFA production was not accompanied by changes in microbiota composition [34]. Although the findings are intriguing, this study has several limitations, including the fact that SCFAs were measured in feces, and not in the proximal colon, the site of their main production, and the fact that it was a cross-sectional sample, that did not control for diet and SCFA absorption. Therefore, these results do not reflect an increased harvesting of energy but only demonstrate an increase in SCFA production/excretion. Contrary to the hypothesis that obesity may be a consequence of increased production and absorption of SCFA, transplanting fecal microbiota from lean subjects into subjects with metabolic syndrome was associated with improved insulin resistance without changes in weight and diet [21]. This study also showed an increase in SCFAs production, specifically butyrate. Butyrate supplementation has been shown to improve glucose metabolism, increase in energy expenditure, and reduction in adiposity in animal models [38]. In addition, propionate has been shown to increase satiety in animals and humans [39–41].
Changes in Metabolic Pathways
Metabolic products of gut microbiota actions can enter the host’s systemic circulation by absorption, enterohepatic circulation, or by a microbiota-induced increase in gut permeability [42]. Although the benefits derived from bacterial production of vitamins and SCFA as an energy source for colonocytes and in colon cancer prevention have long been known, the extensive presence of gut microbial metabolites in our blood stream, and their repercussions for health and disease are just beginning to be appreciated. Metabolomics studies have shown extensive gut microbiota modulation of host systemic metabolic pathways including SCFAs, tryptophan, and tyrosine metabolism [43].
Metabolomic techniques such as nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS) allow for the identification of a large number of molecular metabolites within the biological host system in order to define or fingerprint the functional status of the existing microbiota metabolites to various stimuli [44, 45]. One of the functions of the metabolites in the nutrient-rich environment of the gut is metabolic regulation and offers a unique understanding to the underlying pathophysiology of obesity associated with dietary intake [46]. Below is a summary of various gut metabolites and metabolic pathways involved in influencing obesity.
Changes in Carbohydrate and Lipid Metabolism
The microbiome has been linked to carbohydrate and lipid metabolism. Transplantation of normal mouse microbiota into germ-free mice produced a 60 % increase in body fat content and insulin resistance that seems to be related to an increase in bioavailability of monosaccharides, and the subsequent induction of de novo hepatic lipogenesis. The liver of the conventionalized animals showed an increase in triglyceride content associated with activation of the de novo fatty acid synthesis [47]. Hypertrophy and triglyceride accumulation in the adipocytes were linked to suppression of the fasting-induced adipocyte factor (Fiaf), a circulating lipoprotein lipase inhibitor (LPL), by the conventionalized microbiota resulting in fat storage in white adipose tissue.
Essential amino acids metabolic pathways seem to be associated with obesity and insulin resistance [8•, 25••]. The genes related to these metabolic paths are significantly increased in germ-free mice recipients of obese-twin gut microbiome in comparison to germ-free mice recipients of lean-twin microbiome. The most significantly affected pathways include both essential (phenylalanine, lysine, valine, leucine, and isoleucine) and nonessential (arginine, cysteine, and tyrosine) amino acids [25••]. On the other hand, the transplanted microbiomes of lean twins were richer in genes related to the breakdown and fermentation of diet polysaccharides than the microbiomes of the obese-twin recipients [25••].
Choline is an important part of the cell membrane obtained from the dietary intake of red meat and eggs [48], and is essential for lipid metabolism [49]. Animal [50•] and human [51] studies have shown that microbial activity of dietary choline is associated with altered gut micorbiota composition, which in turn is associated with obesity. The metabolism of dietary choline into trimethylamine-N-oxide (TMAO) has also been correlated with cardiovascular disease and atherosclerosis [52–54], suggesting a strong possible link between dietary intake of choline, gut microbiota, and increased risk for obesity and metabolic disease.
The main bile acids secreted in bile are comprised of cholic acid and chenodeoxycholic acid and are synthesized from cholesterol in order to facilitate the metabolism of dietary fat and the absorption of cholesterol [49]. Five to 10 % of the biotransformation of bile acids takes place by mainly anaerobic gut microbiota (Bacteroides, Eubacterium, and Clostridium), while the rest is secreted in feces. Accordingly, compared to conventionally raised mice, germ-free mice have more bile acid in peripheral tissue but less diverse gut microbiota profiles [55]. Secondary bile acids that are formed from unconjugated free bile acids are actively reabsorbed by bile acid transporters in the ileum and by passive absorption in the large intestine [56, 57]. Bile acids are also signaling molecules that bind to cellular receptors [G-protein-coupled receptor (GPCR) TGR5] that are involved in glucose metabolism [58, 59]. For example, increased TGR5 levels leads to an increase in glucagon-like peptide-1 (GLP-1) levels, which is known to increase glucose tolerance in obese mice [58]. TGR5 also increases the process of energy expenditure and, therefore, protects against diet-related obesity [49, 57, 60]. Bile acids can also activate signaling by binding to nuclear receptors and to GPCRs at the cell surface. The activation of the nuclear receptor FXR stimulates the transcription of genes regulating several metabolic pathways, including bile acid synthesis, cholesterol production, and glucose metabolism and has been associated with improvement in the glucose and lipid profile [61].
Phenols are excreted daily in human feces and in the urine [49]. Although the data on the effect of phenols is still limited, it has been suggested that an increase in phenols can lead to an increased diversity in the composition of gut microbiota (i.e., ratio of Firmicutes versus Bacteroidetes), which can facilitate weight loss [13].
The microbiota of genetically obese mice is rich in enzymes involving the fermentation of dietary fiber including starch/sucrose metabolism, galactose metabolism, and butanoate metabolism [11], and once this obese microbiome is transplanted into germ-free mice, it induces adiposity in their new hosts. This finding was the basis for the microbiome theory related to increased energy harvesting. However, the end products of dietary fiber fermentation include SCFAs such as acetate, propionate, and butyrate [49], which generally improve glucose and energy homeostasis [62]. Butyrate is particularly important in facilitating energy metabolism via cellular metabolism within the colon by the gut microbiota Clostridiales [63]. The importance of the influence of Clostridiales on the increased production of SCFAs was demonstrated in a study where germ-free mice on a high fiber diet were colonized with Clostridiales [64]. These SCFAs serve two important functions in the gut: the suppression of inflammatory immune responses [65, 66] (discussed in detail below) and in the involvement of insulin signaling associated with fat accumulation [67]. SCFAs also modulate the secretion of GLP-1 via the G-protein coupled receptor FFAR2, which is involved in improved insulin secretion [68]. Taken together, these studies suggest that SCFAs are vital gut metabolites involved in the fermentation of dietary fibers and carbohydrates that have important functions in energy metabolism in obesity.
Induction of Low-Grade Inflammation
Systemic inflammatory changes have been identified as a key process in the underlying biological physiology of obesity. For example, a recent review and meta-analysis identified 51 cross-sectional studies investigating the positive correlation (r = 0.36) between obesity in adults and C-reactive protein (a key marker for inflammation) [69]. Similarly, systemic increases in a wide range of inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and adiponectin have also been associated with increasing adiposity [70, 71], and with increased risk for metabolic disorders such as cardiovascular diseases, fatty liver disease, and type 2 diabetes [72, 73]. Despite these various association studies, the causal pathways between obesity, inflammation, and metabolic disease remain incompletely understood. The presence of low-grade systemic inflammation associated with obesity usually involves a complex network of signals interconnecting several organs (e.g., increase in adipokine dysregulation and associated increases in macrophage and lymphocyte recruitment in various tissues) [74, 75]. It is assumed that an increased understanding of the mechanisms driving gut microbiota homeostasis and dysbiosis will lead to a better understanding of the inflammation-related pathophysiology of obesity and consequently could provide an avenue for interventions aimed at modulating gut microbiota in obese individuals [76, 77, 78•, 79].
The mechanisms supporting the influence of disruptions in gut microbiota homeostasis on intestinal inflammation, systemic inflammation, and obesity are still unclear, but the ingestion of high fat diets (HFDs) has been proposed as a possible facilitating factor [78•, 80–83]. Shifts in the gut homeostasis after ingestion of HFD are associated with alterations in the levels and composition of gut microbiota and peptides [76, 77, 78•, 84–86]. It is these diet-induced changes in the micobiota physiology that can cause low-grade systemic inflammation in obesity, and these changes may even precede or predispose one to obesity [76, 77, 78•, 84–86]. Changes in the composition of gut microbiota as a result of increased energy intake can provoke increases in intestinal mucosal inflammation and in changes in gut permeability. These processes together can result in increases in metabolic endotoxemia and in increases of components such as plasma lipopolysaccharides (LPS) within the circulating system [87–89].
Germ-free mice do not show significant increases in body fat despite being fed HFDs, implicating a role of fat induced gut microbiota changes in obesity [19, 47]. On the other hand, when microbiota from conventionally raised mice were transplanted into germ-free mice, increases in body fat were observed [47]. Conventionally raised mice on a Western HFD for 2–16 weeks showed increases in ileal TNF-α mRNA levels and activation of the nuclear factor-kappaB (NF-κB) gene when compared to germ-free mice [80]. Both these inflammatory markers preceded obesity, suggesting the important role of diet-induced changes in the microbiota to promote proinflammatory changes in the gut.
The gut-microbiota-related inflammatory changes leading to obesity following a HFD have been linked to activation of Toll-like receptor 4 (TLR4) signaling and the resulting increase in intestinal levels of LPS [90]. LPS plays a crucial role in the activation of inflammatory and immune processes by binding to lipopolysaccharide-binding proteins and activation of NF-κB pathways [77]. Studies have also shown that increased levels of LPS together with TLR4 are risk factors for obesity [91], insulin resistance [92], and cardiovascular disease [93]. Another Toll-like receptor, TLR5, a key innate trans-membrane protein in the gut mucosa, is involved in the protective process against infection and has also been implicated in obesity related immune activation [94]. For example, a study found that 30 % of mice genetically deficient in TLR5 exhibited colitis, and the other 60 % exhibited 15 % greater body masses compared to their littermates by week 4 [95]. In another study, control mice that were transplanted with feces from TLR5 knockout mice were found to have 20 % increases in body masses, and had epididymal fat pads that were twice as large as those of their control littermates [94]. These fat masses were also correlated with higher serum levels of triglycerides, cholesterol, loss of glycemic control, and with higher levels of blood pressure, which are all characteristics associated with obesity [94]. It is possible that the presence of LPS and TLR4 mediate the presence of obesity in genetically deficient TLR5 mice [81]. This has lead to the hypothesis that alterations in gut microbiota associated with a TLR5 environment facilitates the development of obesity and metabolic disease. In fact, microbiota transplanted from TLR5 knockout mice into germ-free animals exhibited obesity phenotypes including hyperphagia, hyperglycemia, insulin resistance, and increased proinflammatory cytokine levels, suggesting the presence of a low-grade inflammatory mediated obesity [94].
The permeability and integrity of the intestinal mucosa is tightly regulated by membrane and cytoskeletal proteins in the intercellular tight junctions in order to facilitate appropriate absorption and exclusion within the gut [77]. Gut permeability has been implicated as an important factor associated with inflammatory processes in obesity [77, 96]. Other factors contributing to homeostasis and maintenance of the permeability of the gut mucosa include the secretion of mucous and immunoglobulin cells, while proinflammatory cytokines secreted by inflammatory cells cause barrier disruption [97]. Inflammatory processes are known to facilitate destruction of the enteric glial cells, which then leads to the breakdown of the epithelial lining [98]. More recently both preclinical and clinical studies have demonstrated an important role of the gut microbiota in maintaining the integrity of the intestinal epithelium. However, inflammatory processes influence changes in the gut microbiota, which then exacerbate changes related to gut permeability [99–104]. For instance, in addition to the LPS effects on immune activation, high plasma LPS levels have been shown to increase intestinal permeability [105–107]. SCFAs can also play an indirect role on the inflammation-related effects of the gut microbiota on the permeability of the intestine via activation of SCFA receptors on immune cells [77, 108, 109]. Another mechanism involved in effecting the permeability of the intestinal mucosa is the triggering of metabolic endotoxemia [77]. The term metabolic endotoxemia refers to the two- to three-fold increase in intestinal elevations of LPS in healthy controls [81, 110]. However, the long-term effects of metabolic endotoxemia are deleterious and have been linked to metabolic disease such as cardiovascular disease and diabetes [111]. Even though metabolic endotoxemia has been linked to obesity, the exact underlying mechanism has yet to be determined. All these studies show evidence that an increase in gut permeability plays a role in obesity related systemic inflammation.
Effect on Brain and Behaviors Related to Obesity
A growing number of studies have shown that the gut microbiome may influence brain activity and behaviors. For example, several preclinical studies have demonstrated that manipulation of the gut microbiota can alter emotional, nociceptive, and social behaviors (reviewed in Mayer et al. [112•] and Stilling et al. [113]), and produce region specific neurochemical brain changes (reviewed in Cryan and Dinan [114]). Tillisch et al. recently showed that the consumption of a fermented milk probiotic product that contained Bifidobacterium animalis subsp. lactis, Streptococcus thermophiles, Lactobacillus bulgaricus, and Lactococcus lactis subsp. lactis was associated with altered brain responses to an emotion recognition task in healthy volunteers [115].
Regarding obesity, it has been suggested that microbiota could manipulate host behaviors by changing food preferences. For example, altered taste receptors for fat and sweets have been found in germ-free mice [116]. Notably, germ-free mice consumed more sweet solution than wild-type mice, and they displayed an increased number of sweet receptors in the proximal bowel but not in the tongue [116]. In addition, prolonged exposure to high-fat diet results in hyperphagia in animal models. This phenomenon is explained by a decreased activation of vagal afferent neurons [117]. A possible mechanism for this altered activation is the LPS-induced activation of Toll-like receptor 4 (TLR4) on vagal afferent neurons, rendering them insensitive to the effect of leptin and CKK, thus leading to hyperphagia and obesity [82, 118]. In another study, mice lacking TLR5 exhibited an obesity phenotype, features of metabolic syndrome, and hyperphagia. Once fecal matter from the TLR5-deficient mice was transplanted to germ-free mice, similar obesity-related features including hyperphagia were observed [94]. It was hypothesized that the observed hyperphagia resulted from insulin resistance secondary to the gut microbiota-related proinflammatory state, even though other explanations are possible [94]. For example, gut-microbiota-related signaling to the extended reward system has been suggested [119], although experimental data for such a mechanism has not been reported.
High-fat diet feeding has been associated with decreased synthesis of N-acylphosphatidylethanoamide (NAPE) [120]. NAPE is synthesized by the small bowel in response to feeding and is rapidly converted into active N-acylethanolamide (NAE), a family of lipids that decreases food intake in rats and mice [120]. Administration of NAPE by intraperitoneal injection resulted in hypophagia in a dose-dependent fashion that was independent from vagal inervation. Administration of NAPE into the CNS (lateral ventricle) resulted in activation of neurons in the hypothalamus and reduced food consumption [120]. Chen et al. [121] incorporated an engineered NAPE-expressing Escherichia coli bacteria into the gut microbiota by adding it to the drinking water of a DIO mouse model. This intervention was associated with lower food intake, insulin resistance, adiposity, and weight gain, opening the possibility of using engineered bacteria to treat or prevent obesity [121].
Different fermentable carbohydrates have been shown to reduce obesity in animal models [40, 122]. SCFAs, microbial fermentation byproducts, modulate secretion, and gene expression of gut peptides controlling satiety, such as glucagon like peptide-1 (GLP-1) and peptide YY (PYY) by intestinal enteroendocrine cells, suggesting a role for gut microbiota in modulating satiation [39, 65, 68, 123]. Furthermore, probiotics have been associated with increasing numbers of L cells in the intestine and concomitant increased levels of GLP-1 as well as increased sensitivity to Leptin [124]. Most of these mechanisms will actually protect against the development of obesity and its complications. Propionate and butyrate activate intestinal gluconeogenesis via a gut–brain neural circuit involving the fatty acid receptor FFAR3 that improves glucose balance [62]. Moreover, propionate caused neural activation of the dorsal vagal complex and main hypothalamic regions, the paraventricular nucleus (PVN), the lateral hypothalamus (LH), and the arcuate nucleus (ARC) and that activation was prevented by denervation [62]. Dietary manipulation with two fermentable fibers, inulin and β-glucan, resulted in significantly lower body weight gain compared to the mice fed with an HFD without the two added fermentable fibers. Administration of these carbohydrates was associated with an increase in fecal Bifidobacterium and Lactobacillus–Enterococcus. β-Glucan caused a decrease in energy intake and also changes in neuronal signals in the arcuate nucleus, ventromedial hypothalamus, paraventricular nucleus, periventricular nucleus, and the nucleus of the tractus solitarius, suggesting a satiated state [40].
With the exception of bariatric surgery, there are currently no effective treatments for obesity. Gastric bypass is effective in producing weight loss through increases in gut peptides (GLP-1 and PYY), which work in brain centers to produce satiation and reduce food intake [125–128]. Significant changes in gut microbiota have been noted after bariatric surgery, specifically with increases in Proteobacteria (main contributor is Enterobacter hormaechei) and decreases in the Firmicutes and in specific microbiota species (Prevotellaceae and methanogenic Archea) responsible for dietary carbohydrate fermentation and energy harvesting [129]. Gastric bypass produces significant metabolic changes including decreases in fecal bile acids content and increases in production in various amines, which are a reflection of changes in the microbial metabolism of precursors like choline [130••]. The anatomical changes secondary to gastric bypass enhance the colonic microbiota access to partially digested proteins, shifting the protein metabolism to putrefaction. As a result, there is an increase in the production of polyamines such as putrescine and diaminoethane in feces [130••]. Those feces also have increased GABA levels that are associated with microbioal processing of putrescine [130••]. Enterobacteriaceae levels have demonstrated a strong correlation with both postsurgical weight loss and metabolic changes measured as the production of putrescine, uracil, p-cresyl glucuronide, creatinine, and methylamine [130••]. The elevated GABA production by microbial metabolism raises the possibility of brain–gut–microbiome interactions that may play a role in weight loss after gastric bypass.
Conclusions
A large body of evidence supports the view that a change in the metabolic activity of the gut microbiota can contribute to the development of obesity. In contrast, no identifiable group of gut microorganisms have been established that cause or help establish obesity. Converging evidence suggests a complex relationship between the gut microbiome, the host metabolic pathways, immune system, adipose tissue, genetic factors, and the host behaviors and diet. The tight interaction among diet, the gut microbiota, and the host may be the basis of the ancient symbiotic relationship between the microbes and humans. Rapidly advancing analytical technologies have the promise to increase our understanding of the metabolic collaboration between host and gut microbes, and of the mechanisms by which the gut microbiota can influence host ingestive behaviors and immune system responses resulting in obesity.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Obesity and overweight. In: Fact sheet no. 311. March 2011. Accessed 22 Nov 2014.
Collaborators USBoD. The state of US health, 1990–2010: burden of diseases, injuries, and risk factors. JAMA. 2013;310(6):591–608. doi:10.1001/jama.2013.13805.
Sepulveda J, Murray C. The state of global health in 2014. Science. 2014;345(6202):1275–8. doi:10.1126/science.1257099.
Okeke F, Roland BC, Mullin GE. The role of the gut microbiome in the pathogenesis and treatment of obesity. Glob Adv Health Med Improv Healthc Outcomes Worldw. 2014;3(3):44–57. doi:10.7453/gahmj.2014.018.
Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449(7164):811–8. doi:10.1038/nature06245.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi:10.1038/nature08821.
Yang X, Xie L, Li Y, Wei C. More than 9,000,000 unique genes in human gut bacterial community: estimating gene numbers inside a human body. PLoS ONE. 2009;4(6):e6074. doi:10.1371/journal.pone.0006074.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi:10.1038/nature11450. This study used a wide gut metagenome analysis to assess for changes in type-2 diabetes patients; showing that some subsets of bacterial genes are highly specific and may serve as biological markers for type-2 diabetes.
Lederberg J. Infectious history. Science. 2000;288(5464):287–93.
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–48. doi:10.1016/j.cell.2006.02.017.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. doi:10.1038/nature05414.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–3. doi:10.1038/4441022a.
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–5. doi:10.1073/pnas.0504978102.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–4. doi:10.1038/nature07540.
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1(6):6ra14. doi:10.1126/scitranslmed.3000322.
Murphy EF, Cotter PD, Healy S, Marques TM, O’Sullivan O, Fouhy F, et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut. 2010;59(12):1635–42. doi:10.1136/gut.2010.215665.
Woting A, Pfeiffer N, Loh G, Klaus S, Blaut M. Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. mBio. 2014;5(5):e01530-14. doi:10.1128/mBio.01530-14.
Million M, Angelakis E, Maraninchi M, Henry M, Giorgi R, Valero R, et al. Correlation between body mass index and gut concentrations of Lactobacillus reuteri, Bifidobacterium animalis, smithii and Escherichia coli. Int J Obes. 2013;37(11):1460–6. doi:10.1038/ijo.2013.20.
Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104(3):979–84. doi:10.1073/pnas.0605374104.
Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host–archaeal–bacterial mutualism. Proc Natl Acad Sci U S A. 2006;103(26):10011–6. doi:10.1073/pnas.0602187103.
Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143(4):913-6 e7. doi:10.1053/j.gastro.2012.06.031.
Blustein J, Attina T, Liu M, Ryan AM, Cox LM, Blaser MJ, et al. Association of caesarean delivery with child adiposity from age 6 weeks to 15 years. Int J Obes. 2013;37(7):900–6. doi:10.1038/ijo.2013.49.
Bergstrom A, Skov TH, Bahl MI, Roager HM, Christensen LB, Ejlerskov KT, et al. Establishment of intestinal microbiota during early life: a longitudinal, explorative study of a large cohort of Danish infants. Appl Environ Microbiol. 2014;80(9):2889–900. doi:10.1128/AEM. 00342-14.
Graff M, Ngwa JS, Workalemahu T, Homuth G, Schipf S, Teumer A, et al. Genome-wide analysis of BMI in adolescents and young adults reveals additional insight into the effects of genetic loci over the life course. Hum Mol Genet. 2013;22(17):3597–607. doi:10.1093/hmg/ddt205.
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214. doi:10.1126/science.1241214. Elegant experiment controlling for diet and genetic factors to measure the effect of microbiome transplant on phenotype and metabolism. Also shows that phenotype/metabolomic profile can be partially transferred by fecal transplant and points to tight interaction between diet and gut microbiome.
Gaskins HR, Collier CT, Anderson DB. Antibiotics as growth promotants: mode of action. Anim Biotechnol. 2002;13(1):29–42. doi:10.1081/ABIO-120005768.
Petschow B, Dore J, Hibberd P, Dinan T, Reid G, Blaser M, et al. Probiotics, prebiotics, and the host microbiome: the science of translation. Ann N Y Acad Sci. 2013;1306:1–17. doi:10.1111/nyas.12303.
Trasande L, Blustein J, Liu M, Corwin E, Cox LM, Blaser MJ. Infant antibiotic exposures and early-life body mass. Int J Obes. 2013;37(1):16–23. doi:10.1038/ijo.2012.132.
Ajslev TA, Andersen CS, Gamborg M, Sorensen TI, Jess T. Childhood overweight after establishment of the gut microbiota: the role of delivery mode, pre-pregnancy weight and early administration of antibiotics. Int J Obes. 2011;35(4):522–9. doi:10.1038/ijo.2011.27.
Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488(7413):621–6. doi:10.1038/nature11400.
Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81(3):1031–64.
Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40(3):235–43.
Macfarlane GT, Gibson GR, Cummings JH. Comparison of fermentation reactions in different regions of the human colon. J Appl Bacteriol. 1992;72(1):57–64.
Schwiertz A, Taras D, Schafer K, Beijer S, Bos NA, Donus C, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity. 2010;18(1):190–5. doi:10.1038/oby.2009.167.
Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TM, Comelli EM. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes. 2014;4:e121. doi:10.1038/nutd.2014.23.
Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci U S A. 2007;104(25):10643–8. doi:10.1073/pnas.0704189104.
Thauer RK, Jungermann K, Decker K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev. 1977;41(1):100–80.
Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–17. doi:10.2337/db08-1637.
Arora T, Sharma R, Frost G. Propionate. Anti-obesity and satiety enhancing factor? Appetite. 2011;56(2):511–5. doi:10.1016/j.appet.2011.01.016.
Arora T, Loo RL, Anastasovska J, Gibson GR, Tuohy KM, Sharma RK, et al. Differential effects of two fermentable carbohydrates on central appetite regulation and body composition. PLoS ONE. 2012;7(8):e43263. doi:10.1371/journal.pone.0043263.
Vidrine K, Ye J, Martin RJ, McCutcheon KL, Raggio AM, Pelkman C, et al. Resistant starch from high amylose maize (HAM-RS2) and dietary butyrate reduce abdominal fat by a different apparent mechanism. Obesity. 2014;22(2):344–8. doi:10.1002/oby.20501.
Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106(10):3698–703. doi:10.1073/pnas.0812874106.
Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NH, et al. The footprints of gut microbial–mammalian co-metabolism. J Proteome Res. 2011;10(12):5512–22. doi:10.1021/pr2007945.
Nicholson JK. Use of metabolomics to study gut microorganisms. Annu Rev Nutr. 2008. doi:10.1146/annurev-nutr-080508-141243.
Lindon JC, Nicholson JK. Spectroscopic and statistical techniques for information recovery in metabonomics and metabolomics. Annu Rev Anal Chem. 2008;1:45–69. doi:10.1146/annurev.anchem.1.031207.113026.
Xie B, Waters MJ, Schirra HJ. Investigating potential mechanisms of obesity by metabolomics. J Biomed Biotechnol. 2012;2012:805683. doi:10.1155/2012/805683.
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–23. doi:10.1073/pnas.0407076101.
Vance DE. Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr Opin Lipidol. 2008;19(3):229–34. doi:10.1097/MOL.0b013e3282fee935.
Aw W, Fukuda S. Toward the comprehensive understanding of the gut ecosystem via metabolomics-based integrated omics approach. Semin Immunopathol. 2014. doi:10.1007/s00281-014-0456-2.
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–85. doi:10.1038/nature10809. Interesting study on interaction between inflammatory response and gut microbiome composition and its role in progression of fatty liver disease and obesity, of note similar findings were seen when altered microbiome was transmitted via cohabitation.
Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140(3):976–86. doi:10.1053/j.gastro.2010.11.049.
Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi:10.1038/nature09922.
Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A. 2006;103(33):12511–6. doi:10.1073/pnas.0601056103.
Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–85. doi:10.1038/nm.3145.
Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4523–30. doi:10.1073/pnas.1006734107.
Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–59. doi:10.1194/jlr. R500013-JLR200.
Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–9. doi:10.1038/nature04330.
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–77. doi:10.1016/j.cmet.2009.08.001.
Prawitt J, Abdelkarim M, Stroeve JH, Popescu I, Duez H, Velagapudi VR, et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes. 2011;60(7):1861–71. doi:10.2337/db11-0030.
Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci U S A. 2008;105(36):13580–5. doi:10.1073/pnas.0804437105.
Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006;103(4):1006–11. doi:10.1073/pnas.0506982103.
De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell. 2014;156(1–2):84–96. doi:10.1016/j.cell.2013.12.016.
Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13(5):517–26. doi:10.1016/j.cmet.2011.02.018.
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504(7480):446–50. doi:10.1038/nature12721.
Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A. 2008;105(43):16767–72. doi:10.1073/pnas.0808567105.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6. doi:10.1038/nature08530.
Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi:10.1038/ncomms2852.
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012;61(2):364–71. doi:10.2337/db11-1019.
Choi J, Joseph L, Pilote L. Obesity and C-reactive protein in various populations: a systematic review and meta-analysis. Obes Rev Off J Int Assoc Study Obes. 2013;14(3):232–44. doi:10.1111/obr.12003.
Marques-Vidal P, Bochud M, Bastardot F, Luscher T, Ferrero F, Gaspoz JM, et al. Association between inflammatory and obesity markers in a Swiss population-based sample (CoLaus Study). Obes Facts. 2012;5(5):734–44. doi:10.1159/000345045.
Bahceci M, Gokalp D, Bahceci S, Tuzcu A, Atmaca S, Arikan S. The correlation between adiposity and adiponectin, tumor necrosis factor alpha, interleukin-6 and high sensitivity C-reactive protein levels. Is adipocyte size associated with inflammation in adults? J Endocrinol Investig. 2007;30(3):210–4.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7. doi:10.1038/nature05485.
Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. 2006;83(2):461S–5S.
Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314(1):1–16. doi:10.1016/j.mce.2009.07.031.
Sell H, Eckel J. Adipose tissue inflammation: novel insight into the role of macrophages and lymphocytes. Curr Opin Clin Nutr Metab Care. 2010;13(4):366–70. doi:10.1097/MCO.0b013e32833aab7f.
Sanz Y, Moya-Perez A. Microbiota, inflammation and obesity. Adv Exp Med Biol. 2014;817:291–317. doi:10.1007/978-1-4939-0897-4_14.
Cox AJ, West NP, Cripps AW. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2014. doi:10.1016/S2213-8587(14)70134-2.
Bleau C, Karelis AD, St-Pierre DH, Lamontagne L. Crosstalk between intestinal microbiota, adipose tissue and skeletal muscle as an early event in systemic low grade inflammation and the development of obesity and diabetes. Diabetes Metab Res Rev. 2014. doi:10.1002/dmrr.2617. Good review of interactions between gut microbiome, intestinal barrier and inflammation in obesity development.
Peterson CT, Sharma V, Elmen L, Peterson SN. Immune homeostasis, dysbiosis and therapeutic modulation of the gut microbiota. Clin Exp Immunol. 2014. doi:10.1111/cei.12474.
Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, et al. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE. 2010;5(8):e12191. doi:10.1371/journal.pone.0012191.
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–81. doi:10.2337/db07-1403.
de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;299(2):G440–8. doi:10.1152/ajpgi.00098.2010.
Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2010;59(1):172–81. doi:10.2337/db09-0367.
Gangarapu V, Yildiz K, Ince AT, Baysal B. Role of gut microbiota: obesity and NAFLD. Turk J Gastroenterol. 2014;25(2):133–40. doi:10.5152/tjg.2014.7886.
Moran CP, Shanahan F. Gut microbiota and obesity: role in aetiology and potential therapeutic target. Best Pract Res Clin Gastroenterol. 2014;28(4):585–97. doi:10.1016/j.bpg.2014.07.005.
Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger AG. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benefic Microbes. 2014;5(1):33–43. doi:10.3920/BM2013.006.
Raybould HE. Gut microbiota, epithelial function and derangements in obesity. J Physiol. 2012;590(Pt 3):441–6. doi:10.1113/jphysiol.2011.222133.
Shen J, Obin MS, Zhao L. The gut microbiota, obesity and insulin resistance. Mol Asp Med. 2013;34(1):39–58. doi:10.1016/j.mam.2012.11.001.
Little TJ, Feinle-Bisset C. Effects of dietary fat on appetite and energy intake in health and obesity—oral and gastrointestinal sensory contributions. Physiol Behav. 2011;104(4):613–20. doi:10.1016/j.physbeh.2011.04.038.
Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE. 2012;7(10):e47713. doi:10.1371/journal.pone.0047713.
Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56(8):1986–98. doi:10.2337/db06-1595.
Song MJ, Kim KH, Yoon JM, Kim JB. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun. 2006;346(3):739–45. doi:10.1016/j.bbrc.2006.05.170.
Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci U S A. 2004;101(29):10679–84. doi:10.1073/pnas.0403249101.
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328(5975):228–31. doi:10.1126/science.1179721.
Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117(12):3909–21. doi:10.1172/JCI33084.
Moreno-Navarrete JM, Sabater M, Ortega F, Ricart W, Fernandez-Real JM. Circulating zonulin, a marker of intestinal permeability, is increased in association with obesity-associated insulin resistance. PLoS ONE. 2012;7(5):e37160. doi:10.1371/journal.pone.0037160.
Kurashima Y, Goto Y, Kiyono H. Mucosal innate immune cells regulate both gut homeostasis and intestinal inflammation. Eur J Immunol. 2013;43(12):3108–15. doi:10.1002/eji.201343782.
Yu YB, Li YQ. Enteric glial cells and their role in the intestinal epithelial barrier. World J Gastroenterol. 2014;20(32):11273–80. doi:10.3748/wjg.v20.i32.11273.
Ewaschuk JB, Diaz H, Meddings L, Diederichs B, Dmytrash A, Backer J, et al. Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am J Physiol Gastrointest Liver Physiol. 2008;295(5):G1025–34. doi:10.1152/ajpgi.90227.2008.
Anderson RC, Cookson AL, McNabb WC, Kelly WJ, Roy NC. Lactobacillus plantarum DSM 2648 is a potential probiotic that enhances intestinal barrier function. FEMS Microbiol Lett. 2010;309(2):184–92. doi:10.1111/j.1574-6968.2010.02038.x.
Shen TY, Qin HL, Gao ZG, Fan XB, Hang XM, Jiang YQ. Influences of enteral nutrition combined with probiotics on gut microflora and barrier function of rats with abdominal infection. World J Gastroenterol. 2006;12(27):4352–8.
Ukena SN, Singh A, Dringenberg U, Engelhardt R, Seidler U, Hansen W, et al. Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PLoS ONE. 2007;2(12):e1308. doi:10.1371/journal.pone.0001308.
Karczewski J, Troost FJ, Konings I, Dekker J, Kleerebezem M, Brummer RJ, et al. Regulation of human epithelial tight junction proteins by Lactobacillus plantarum in vivo and protective effects on the epithelial barrier. Am J Physiol Gastrointest Liver Physiol. 2010;298(6):G851–9. doi:10.1152/ajpgi.00327.2009.
Lamprecht M, Bogner S, Schippinger G, Steinbauer K, Fankhauser F, Hallstroem S, et al. Probiotic supplementation affects markers of intestinal barrier, oxidation, and inflammation in trained men; a randomized, double-blinded, placebo-controlled trial. J Int Soc Sports Nutr. 2012;9(1):45. doi:10.1186/1550-2783-9-45.
Li Q, Zhang Q, Wang C, Liu X, Li N, Li J. Disruption of tight junctions during polymicrobial sepsis in vivo. J Pathol. 2009;218(2):210–21. doi:10.1002/path.2525.
Lai CW, Sun TL, Lo W, Tang ZH, Wu S, Chang YJ, et al. Shedding-induced gap formation contributes to gut barrier dysfunction in endotoxemia. J Trauma Acute Care Surg. 2013;74(1):203–13. doi:10.1097/TA.0b013e3182788083.
Jorgensen VL, Nielsen SL, Espersen K, Perner A. Increased colorectal permeability in patients with severe sepsis and septic shock. Intensive Care Med. 2006;32(11):1790–6. doi:10.1007/s00134-006-0356-6.
Covington DK, Briscoe CA, Brown AJ, Jayawickreme CK. The G-protein-coupled receptor 40 family (GPR40-GPR43) and its role in nutrient sensing. Biochem Soc Trans. 2006;34(Pt 5):770–3. doi:10.1042/BST0340770.
Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278(28):25481–9. doi:10.1074/jbc.M301403200.
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. doi:10.2337/db06-1491.
Kallio KA, Hatonen KA, Lehto M, Salomaa V, Mannisto S, Pussinen PJ. Endotoxemia, nutrition, and cardiometabolic disorders. Acta Diabetol. 2014. doi:10.1007/s00592-014-0662-3.
Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci Off J Soc Neurosci. 2014;34(46):15490–6. doi:10.1523/JNEUROSCI. 3299-14.2014. Comprehensive review on current evidence on interaction between gut microbiome and brain function.
Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour—epigenetic regulation of the gut-brain axis. Genes Brain Behav. 2014;13(1):69–86. doi:10.1111/gbb.12109.
Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701–12. doi:10.1038/nrn3346.
Tillisch K, Labus J, Kilpatrick L, Jiang Z, Stains J, Ebrat B, et al. Consumption of fermented milk product with probiotic modulates brain activity. Gastroenterology. 2013;144(7):1394–401. doi:10.1053/j.gastro.2013.02.043. 401 e1-4.
Swartz TD, Duca FA, de Wouters T, Sakar Y, Covasa M. Up-regulation of intestinal type 1 taste receptor 3 and sodium glucose luminal transporter-1 expression and increased sucrose intake in mice lacking gut microbiota. Br J Nutr. 2012;107(5):621–30. doi:10.1017/S0007114511003412.
Covasa M. Deficits in gastrointestinal responses controlling food intake and body weight. Am J Physiol Regul Integr Comp Physiol. 2010;299(6):R1423–39. doi:10.1152/ajpregu.00126.2010.
de Lartigue G, Barbier de la Serre C, Espero E, Lee J, Raybould HE. Diet-induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab. 2011;301(1):E187–95. doi:10.1152/ajpendo.00056.2011.
Alcock J, Maley CC, Aktipis CA. Is eating behavior manipulated by the gastrointestinal microbiota? Evolutionary pressures and potential mechanisms. Bioessays. 2014;36(10):940–9. doi:10.1002/bies.201400071.
Gillum MP, Zhang D, Zhang XM, Erion DM, Jamison RA, Choi C, et al. N-acylphosphatidylethanolamine, a gut- derived circulating factor induced by fat ingestion, inhibits food intake. Cell. 2008;135(5):813–24. doi:10.1016/j.cell.2008.10.043.
Chen Z, Guo L, Zhang Y, Walzem RL, Pendergast JS, Printz RL, et al. Incorporation of therapeutically modified bacteria into gut microbiota inhibits obesity. J Clin Invest. 2014;124(8):3391–406. doi:10.1172/JCI72517.
So PW, Yu WS, Kuo YT, Wasserfall C, Goldstone AP, Bell JD, et al. Impact of resistant starch on body fat patterning and central appetite regulation. PLoS ONE. 2007;2(12):e1309. doi:10.1371/journal.pone.0001309.
Lin HV, Frassetto A, Kowalik Jr EJ, Nawrocki AR, Lu MM, Kosinski JR, et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE. 2012;7(4):e35240. doi:10.1371/journal.pone.0035240.
Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes. 2011;60(11):2775–86. doi:10.2337/db11-0227.
Scholtz S, Miras AD, Chhina N, Prechtl CG, Sleeth ML, Daud NM, et al. Obese patients after gastric bypass surgery have lower brain-hedonic responses to food than after gastric banding. Gut. 2014;63(6):891–902. doi:10.1136/gutjnl-2013-305008.
Peterli R, Steinert RE, Woelnerhanssen B, Peters T, Christoffel-Courtin C, Gass M, et al. Metabolic and hormonal changes after laparoscopic Roux-en-Y gastric bypass and sleeve gastrectomy: a randomized, prospective trial. Obes Surg. 2012;22(5):740–8. doi:10.1007/s11695-012-0622-3.
Basso N, Capoccia D, Rizzello M, Abbatini F, Mariani P, Maglio C, et al. First-phase insulin secretion, insulin sensitivity, ghrelin, GLP-1, and PYY changes 72 h after sleeve gastrectomy in obese diabetic patients: the gastric hypothesis. Surg Endosc. 2011;25(11):3540–50. doi:10.1007/s00464-011-1755-5.
le Roux CW, Welbourn R, Werling M, Osborne A, Kokkinos A, Laurenius A, et al. Gut hormones as mediators of appetite and weight loss after Roux-en-Y gastric bypass. Ann Surg. 2007;246(5):780–5. doi:10.1097/SLA.0b013e3180caa3e3.
Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A. 2009;106(7):2365–70. doi:10.1073/pnas.0812600106.
Li JV, Ashrafian H, Bueter M, Kinross J, Sands C, le Roux CW, et al. Metabolic surgery profoundly influences gut microbial–host metabolic cross-talk. Gut. 2011;60(9):1214–23. doi:10.1136/gut.2010.234708. This study provides a comprehensive description of changes in gut anatomy, microbiota profile and metabolism after gastric bypass and possible explanations of their influence in weight loss after gastric bypass.
Acknowledgments
This study was supported in part by the National Institutes of Health grants R01 DK048351 (EAM), P50 DK064539 (EAM), and P30 DK041301. The authors thank Ms. Cathy Liu for invaluable editorial and graphic design assistance.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Claudia Sanmiguel, Arpana Gupta, and Emeran A. Mayer declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
The human studies contained in this article that were perfomed by any of the authors were IRB approved and subjects were consented before their participation in the study.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Obesity Treatment
Rights and permissions
About this article

Cite this article
Sanmiguel, C., Gupta, A. & Mayer, E.A. Gut Microbiome and Obesity: A Plausible Explanation for Obesity. Curr Obes Rep 4, 250–261 (2015). https://doi.org/10.1007/s13679-015-0152-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-015-0152-0