
Abstract
Acute exacerbation of idiopathic pulmonary fibrosis (IPF) occurs in approximately 10 % of IPF patients annually, and is a leading cause of morbidity and mortality in this disease. Although currently defined as idiopathic acute worsening, acute exacerbation of IPF may in fact have a variety of causes, in particular infection and aspiration. Central to the pathobiology of clinically meaningful events is a diffuse injury to the IPF lung, manifest histopathologically as diffuse alveolar damage, and biologically as accelerated alveolar epithelial cell injury or repair. On the basis of these recent observations, we propose a new conceptual framework for acute exacerbation of IPF that removes the idiopathic requirement and focuses on the pathophysiological mechanism involved.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease of unknown cause. The reported median survival for patients with IPF is approximately three years from the time of diagnosis, and there is no clearly effective therapy that improves survival [1••]. IPF patients experience episodes of acute respiratory worsening that result in substantial morbidity and mortality [1••, 2, 3••]. Although known causes of acute respiratory worsening, for example pneumonia, heart failure and thromboembolism, account for a proportion of these episodes, many remain idiopathic after clinical review. Idiopathic episodes of acute respiratory worsening have been termed acute exacerbations of IPF [2, 4]. This paper will review the current understanding of acute exacerbations of IPF (AE-IPF) and propose a new conceptual framework for thinking about acute exacerbation in IPF patients.
Definitions
Acute exacerbation of IPF was first defined by Kondo, in 1989, as an acute, clinically significant respiratory worsening of unidentifiable cause, in a patient with underlying IPF [5]. Kondoh et al. further developed this concept [4]. Restricting diagnosis of acute exacerbation of IPF to idiopathic events distinguished IPF from other chronic lung diseases, e.g. asthma and chronic obstructive pulmonary disease, where definitions of acute exacerbation are not limited to idiopathic cases.
In 2007, the IPF Clinical Research Network (IPFnet) proposed diagnostic criteria for AE-IPF on the basis of the criteria of Kondo, Kondoh, and others (Table 1) [2]. Acute exacerbation was defined by: subjective worsening of dyspnea within the prior 30 days; new bilateral opacities on high-resolution computed tomography (HRCT) of the chest; no evidence of infection on endotracheal or bronchoalveolar lavage analysis; and the exclusion of other causes. Patients with idiopathic respiratory worsening who failed to meet all of the above criteria were diagnosed with suspected acute exacerbation of IPF.
These diagnostic criteria have enhanced our understanding of a previously under-appreciated aspect of the natural history of IPF. Fundamental to their adoption, however, is the assumption that idiopathic acute respiratory worsening in IPF is a distinct clinical entity that should be distinguished from acute respiratory worsening of known cause. That is, AE-IPF is assumed to have a unique cause or pathobiology that has therapeutic or prognostic implications. In our opinion, data published over the last several years questions the validity of this assumption. As we will discuss below, we believe this has implications for our understanding of acute exacerbation of IPF.
Incidence
Acute exacerbation of IPF occurs with an estimated incidence of 5–15 % per year, with lower estimates typically arising from the placebo cohorts of clinical trials [6–9] and higher estimates from observational cohorts [3••, 10, 11]. This variability is probably attributable to differences in patient characteristics (in particular severity of physiological impairment) between cohorts, and to the logistical challenges of identifying AE-IPF in multicenter clinical trials.
Risk factors
Several risk factors for AE-IPF have been suggested, including reduced baseline lung function (in particular reduced forced vital capacity (FVC)), increased dyspnea, a history of coronary artery disease, and pulmonary hypertension [3••, 11–13]. These suggest that acute exacerbation of IPF is more common in patients with more advanced disease. Prednisone use has been associated with an increased risk of AE-IPF in retrospective cohorts; however, it is difficult to exclude confounding by indication (i.e. that patients were getting prednisone because of the severity of their disease or because they were experiencing exacerbation) [3••, 13]. Never having smoked cigarettes has been associated with an increased acute exacerbation risk [3••]. There seems to be a seasonal pattern to AE-IPF, with increased frequency of exacerbation during the winter months [12–14]. Patients with a history of AE-IPF have more than four times the risk of future AE-IPF events [15]. Other descriptive cohorts have identified subsets of patients who have experienced multiple AE-IPF over time, but it is unclear whether this phenomenon arises from patient-specific or environmental factors.
Clinical presentation
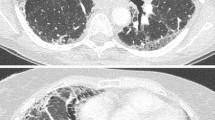
Acute exacerbation of IPF can occur at any point during the course of IPF and may be the presenting manifestation of disease. When the latter occurs, characteristic radiography and pathology findings are essential for diagnosis. Patients with acute exacerbation typically present with symptoms of worsening dyspnea, cough and fever [1••, 2]. By definition, AE-IPF is associated with new bilateral radiographic opacities on HRCT that can be peripheral, multifocal or diffuse in distribution [16]. The pattern of radiographic involvement has prognostic implications (see below).
Surgical lung biopsy of patients with AE-IPF usually identifies acute and organizing diffuse alveolar damage (DAD), with a predominance of organizing pneumonia, and extensive fibroblastic foci occasionally observed [17, 18]. Whether the latter indicate sampling error is unknown. In most cases, dense, established fibrosis with or without microscopic honeycombing will also be present [19]. These findings, and exuberant fibroblastic foci, distinguish AE-IPF from isolated DAD.
Diagnostic work-up
There is no standardized work-up for patients presenting with possible AE-IPF, but testing often focuses on trying to identify a treatable underlying cause of respiratory worsening, for example infection, pulmonary embolism, or congestive heart failure. Computed tomography-angiography is very useful for evaluating most of these possibilities and should be performed for all patients. The necessity of bronchoalveolar lavage in testing for infectious agents is less clear to many clinicians, because the sensitivity of most microbiological tests is poor; empirical therapy is often started regardless. There is also a risk that bronchoscopy will worsen the hypoxemia of non-intubated patients with high baseline oxygen requirements; a sputum or nasopharyngeal swab can almost always be sent in these cases. For intubated patients, a bronchoalveolar lavage or an endotracheal aspirate can usually be obtained safely and easily. We believe use of surgical lung biopsy for an established IPF patient with possible AE-IPF should be discouraged, because of the high risk of peri-operative morbidity and mortality and the low probability of altered management or prognosis.
Etiology of acute exacerbation of IPF
The most commonly stated etiological hypothesis for AE-IPF is that it is an acute, intrinsic acceleration of IPF disease progression. It is also possible that AE-IPF indicates clinically occult secondary events, for example infection, aspiration or heart failure. Recent studies have attempted to investigate the etiology of AE-IPF by describing the biological phenotype of AE-IPF and looking more rigorously for potential causes. These studies suggest that AE-IPF is indeed an acceleration of the IPF disease process, but that it is probably caused by occult secondary insults, for example infection, aspiration and mechanical stress.
Biological phenotype of acute exacerbation of IPF
In 2009, Konishi et al. published a study of gene expression profiles of lungs from recently deceased patients with AE-IPF or non-exacerbated IPF, and from non-diseased controls [20]. Compared with controls, AE-IPF and non-exacerbated IPF profiles were similar. Interestingly, no changes to inflammation-associated genes were identified. Instead, AE-IPF was primarily characterized by evidence of enhanced epithelial cell activity (injury and/or proliferation). Subsequently, our group in collaboration with investigators in Korea analyzed blood biomarker profiles of AE-IPF, stable IPF and acute lung injury (ALI) [21•]. Consistent with the data of Konishi, biomarkers of type II alveolar epithelial cell activity were elevated for AE-IPF compared with stable IPF and ALI, and levels of inflammatory biomarkers were normal. Kakugawa et al. revealed elevated levels of heat shock 47 proteins (HSP47) for AE-IPF compared with stable IPF [22]. HSP47 is a collagen-specific molecular chaperone necessary for collagen synthesis and deposition, and has been revealed to positively correlate with IPF survival [23]. Together, these results suggest that AE-IPF, once established, is characterized by an acceleration of the underlying IPF disease (i.e. an alveolar epithelial-cell-induced fibroproliferative process).
Potential causes of acute exacerbation of IPF
Infection
Huie et al. investigated the potential role of infection in acute respiratory worsening for 27 patients with fibrotic lung disease, 13 of whom had IPF [24]. For eight cases, a potential infectious etiology was identified by use of standard culture-based techniques and polymerase chain reaction (PCR) or antigen-based testing for common respiratory viruses. Of these eight, three had bacterial pneumonia, one had Pneumocystis jirovecii pneumonia, and four had evidence of replicating virus (two herpes simplex virus, one cytomegalovirus, and one parainfluenza virus).
Building on these results, our group, again in collaboration with investigators in Korea and Japan, used genomics-based techniques to investigate the role of viral infection as a potential cause of AE-IPF [25•]. Bronchoalveolar lavage (BAL) fluid from patients with AE-IPF was compared with that of patients with stable IPF by use of a pan-viral microarray and, for a subset, deep sequencing. Four of 43 AE-IPF samples (9 %) were positive for common respiratory viruses (two rhinovirus, one coronovirus, one parainfluenza virus) compared with none from the stable-IPF group. Fifteen additional viruses were identified in AE-IPF samples (one herpes simplex virus, two Epstein–Barr virus, and twelve torque teno virus), whereas no additional viruses were detected in the stable IPF group samples. The clinical significance of these non-respiratory viruses was unclear. Respiratory viruses may have been undetectable in some additional samples because of a delay between the onset of symptoms and BAL collection. Collectively, these data suggest that some AE-IPF cases are caused by clinically occult viral infection.
Aspiration of gastric contents
Abnormal gastroesophageal reflux (GER) is almost universal in IPF and is a known risk factor for aspiration [26•, 27, 28]. Two small studies of patients with GER and IPF have suggested stabilization both of the disease and of oxygenation by medical or surgical treatment of GER [29, 30]. Our group, in collaboration with investigators from Korea, measured pepsin levels (a biomarker of aspiration) in the BAL fluid of IPF patients with and without acute exacerbation [26•]. For this cohort, BAL pepsin level was positively associated with acute exacerbation status, a finding mostly resulting from the presence of a subgroup of AE-IPF cases (33 %) whose BAL pepsin levels were substantially elevated. A recent study, comparing outcomes for patients on and off proton pump inhibitor (PPI) therapy for abnormal GER, found PPI therapy to be associated with significantly reduced incidence of AE-IPF [31•]. These data suggest that aspiration of gastric contents, in particular acid, may be a cause of AE-IPF for a subgroup of patients.
Mechanical stress
There is increasing evidence that surgery can cause acute respiratory worsening in IPF, presumably the result of increased mechanical stress to the lungs. A large cohort of AE-IPF patients included 15 with a post-operative onset (eight lung biopsies, three lung resections and three non-respiratory surgeries) [3••]. Prolonged mechanical ventilation, high tidal volume and high concentration of supplemental oxygen during surgery have been proposed as potential causes [32, 33]. Other contributors to increased risk of AE-IPF after surgery may be the presence of typical histological honeycombing [34], increased extent of radiographic fibrosis [35], elevated serum Krebs von den Lungen-6 (KL-6), greater extent of surgical resection (lobe vs. wedge resection) [36], high intra-operative fluid balance, and pre-operative elevation of C-reactive protein (CRP) [37]. There may also be an increased risk of AE-IPF post-BAL, particularly for IPF patients with poor lung function undergoing multiple BAL procedures [38, 39].
Ambient air pollution
Finally, there is recent evidence that ambient air pollution exposure, notably ozone (O3) and nitrogen dioxide (NO2), may cause AE-IPF in some patients with IPF. In a retrospective analysis of a large well-defined cohort of IPF patients, our group in collaboration with investigators in Korea found that the mean and maximum exposures to O3 and NO2 were positively associated with an increased risk of AE-IPF [15]. Additionally, increased frequency of exposure to pollution levels exceeding published air quality standards was associated with an increased risk of AE-IPF.
Management
Prevention
Prevention of AE-IPF may prove the most effective approach to management (Table 2); there are currently no data from which to assess efficacy. Annual influenza vaccination (and pneumococcal vaccination where indicated) makes intuitive sense as a potential way to protect against respiratory infections that could cause acute exacerbation. Hand washing and other infection control policies are also prudent. Behavioral measures to reduce the risk of microaspiration (small meals, adequate time between meals and bed, elevation of the head of the bed) may also be appropriate general preventive measures for IPF patients. For IPF patients with documented abnormal GER, it may also be appropriate to consider medical or surgical treatment of GER to reduce the risk of aspiration-induced AE-IPF. For patients with IPF who are undergoing surgery, reducing the partial pressure of oxygen and tidal volumes used intra-operatively, when possible, may help reduce the risk of mechanical stress causing AE-IPF. Use of air quality measures (choice of living environment, and minimizing exposure to airborne irritants and pollutants including ambient ozone and nitrogen dioxide) may also be appropriate. Finally, therapy with the objective of treating the underlying fibroproliferation of IPF may also reduce the risk of AE-IPF, by controlling the pathological response of the IPF lung to potential stressors [8].
Therapy
Once a patient with IPF develops an acute exacerbation, management is largely supportive (Table 2). Many patients with AE-IPF present with respiratory failure, and the decision to intubate and mechanically ventilate is a crucial one. A retrospective study of 24 IPF patients identified eight admitted to the ICU during AE-IPF, and found that 22/24 died during hospital admission, with none of the AE patients surviving to discharge [40]. In another retrospective study, 24/25 IPF patients admitted to the ICU for respiratory failure of unknown cause died in hospital despite thorough work-up and corticosteroid therapy [41]. In the largest cohort study of AE-IPF published to date, approximately 50 % of patients required ICU admission, and 80 % of these patients died during the follow-up period (not necessarily during the hospitalization) [3••]. On the basis of these data, many IPF patients will decide against intubation. Ideally, this decision should be made with the patient and family, well ahead of time and after a thoughtful and measured discussion.
Most patients with AE-IPF receive corticosteroids, in accordance with the weak positive recommendation of the 2011 international evidence-based guidelines [1••]. There have been no randomized trials investigating the use of corticosteroids in AE-IPF. The rationale for corticosteroid use in AE-IPF is twofold. First, for a minority of cases of AE-IPF organizing pneumonia has been revealed on surgical lung biopsy, suggesting a corticosteroid-responsive element. Second, some data support the use of late steroids for acute lung injury (although this is a contentious topic in the acute lung injury community), and acute lung injury has diffuse alveolar damage on surgical lung biopsy similar to that of AE-IPF. It is unknown what dose and duration of corticosteroids to use. For AE-IPF patients with respiratory failure, our practice is to treat with high-dose intravenous corticosteroids (e.g. 500 mg solumedrol daily) for 3–5 days, with tapering to a lower dose or discontinuation on the basis of clinical response. For less severe cases, oral prednisone 60 mg daily is used for 10–14 days with tapering as clinically indicated. Careful attention must be given to the individual risks of corticosteroid treatment for AE-IPF patients.
Most patients presenting with AE-IPF receive empirical antibiotics targeting respiratory pathogens, although there is no data to support their use [42]. This is on the basis that underlying infection can be easily missed by microbiological testing, and that a treatment course of antimicrobial therapy is low risk. Our practice is to treat patients empirically for common community-acquired organisms.
Several small studies have reported varying efficacy of novel therapy for AE-IPF, but none has been rigorously tested in large randomized trials. Experimental therapy includes combined tacrolimus and corticosteroids [43], polymyxin B-immobilized fiber column perfusion treatment [44–48], cyclosporine-A [49, 50] and recombinant thrombomodulin [51].
Prognosis
Short-term survival post-AE-IPF is poor, with a median survival of approximately two months post-event [3••]. Several markers of worse prognosis for AE-IPF have been identified, including lower baseline FVC and DLCO, elevated lactate dehydrogenase and CRP, and higher body mass index [3••, 12, 52]. Circulating fibrocytes and elevated Interleukin-17 levels may also be prognostic [45, 53]. Unsurprisingly, worsening gas-exchange despite treatment is a poor prognostic marker [12]. The radiographic pattern of involvement at the time of AE-IPF is also prognostic, with higher mortality associated with diffuse ground-glass abnormality compared with a multifocal or peripheral distribution [16]. A composite HRCT score, including extent of ground-glass opacification, consolidation, traction bronchiectasis, and honeycombing, revealed that higher scores predicted increased mortality risk after AE-IPF [54]. Such prognostic information may be useful for appropriate triage, and for decision-making regarding level and continuation of care.
The paucity of data on outcomes after lung transplantation currently precludes a recommendation for or against lung transplantation for patients experiencing AE-IPF. Individual outcomes probably depend on a combination of patient factors and center-specific experience.
Proposed conceptual framework
Defining AE-IPF has catalyzed the recognition and characterization of a previously under-recognized and under-appreciated aspect of the natural history of IPF. We believe the current definition, however, has led to an organizational framework for acute respiratory worsening in IPF that emphasizes and prioritizes idiopathic events over those of known cause—a framework that emerging data suggest may be conceptually flawed.
Data reviewed earlier in this article suggest that many so-called acute exacerbations of IPF are in fact not idiopathic. Instead they seem to be caused by a variety of secondary events, including infection and aspiration. In support of this hypothesis, epidemiological data reveal that AE-IPF and acute respiratory worsening of known cause have similar clinical characteristics and outcomes [3••, 13]. Taken together, the above data suggest that AE-IPF as currently defined may be an artificial construct—a clinical distinction without biological or clinical evidence to support it. This hypothesis could of course be wrong, but we believe the current balance of evidence argues in its favor.
This does not mean that the concept of acceleration of the underlying disease in AE-IPF is incorrect. Indeed, microarray and biomarker data reviewed earlier suggest that the overall evidence from AE-IPF cases does indeed indicate accelerated disease. We speculate that for patients with AE-IPF, unrelated clinical events (e.g. infection, microaspiration), that in non-diseased lungs would perhaps cause no clinically significant sequelae, lead to diffuse alveolar injury and acceleration of the fibroproliferative process. The IPF lung may be prone to sudden decline because of its inability to undergo normal alveolar repair, and may require only minor stressors to initiate a rapid decline.
In conclusion, we propose that clinicians and researchers consider a new conceptual framework for acute exacerbation in IPF that de-emphasizes etiology (i.e. removes the idiopathic requirement) and emphasizes the presence of diffuse injury signifying clinically significant disease worsening (Figure 1). In this conceptual model, the hypothesis is that all acute respiratory worsening of IPF have a secondary cause (some clinically identifiable, some not), and that some acute respiratory worsening lead to acceleration of the underlying disease, resulting in diffuse alveolar injury—i.e. exacerbation. We encourage researchers to think creatively about AE-IPF and consider alternative conceptual frameworks, for example the one presented here. More research into AE-IPF is needed to further improve our understanding of these clinically meaningful events.

Proposed conceptual framework for acute exacerbation of idiopathic pulmonary fibrosis (IPF). Patients with IPF and accelerated worsening of dyspnea can have parenchymal or extra-parenchymal explanations for their symptoms. We propose that those with parenchymal causes be categorized as acute respiratory worsening. Cases of acute respiratory worsening with evidence of diffuse alveolar injury on high-resolution computed tomography (HRCT) scanning are subsequently categorized as acute exacerbation of IPF. For some cases of acute exacerbation of IPF, a cause will be identified (e.g. infection). Cases of acute respiratory worsening with no HRCT evidence for diffuse alveolar injury are categorized as other acute respiratory worsening, possible explanations for which are bronchitis, lobar pneumonia, and sub-radiographic acute exacerbation of IPF. PE, pulmonary embolism; CHF, congestive heart failure; PTX, pneumothorax
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Raghu G et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. These are the most recent internationally accepted consensus guidelines on the diagnosis and management of IPF.
Collard HR et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(7):636–43.
•• Song JW et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37(2):356–63. This manuscript describes clinical features and outcomes in acute exacerbations of IPF for the largest descriptive cohort to date. It also reveals that acute exacerbation may occur after surgery.
Kondoh Y et al. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest. 1993;103(6):1808–12.
Kondo A, S.S. Acute exacerbation in idiopathic interstitial pneumonia (IIP). Intractable Diseases Research Foundation Publication No. 27. ed. Interstitial pneumonia of unknown etiology. 1989, Tokyo, Japan.: University of Tokyo Press.
King Jr TE et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–9.
Noble PW et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
Richeldi L et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–87.
Zisman DA et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–8.
Mura M et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J. 2012;40(1):101–9.
Judge EP et al. Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Eur Respir J. 2012;40(1):93–100.
Simon-Blancal V et al. Acute exacerbation of idiopathic pulmonary fibrosis: outcome and prognostic factors. Respiration. 2012;83(1):28–35.
Collard HR et al. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res. 2013;14:73.
Olson AL et al. Seasonal variation: mortality from pulmonary fibrosis is greatest in the winter. Chest. 2009;136(1):16–22.
Johannson KA et al. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur Respir J. (in press).
Akira M et al. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(4):372–8.
Parambil JG, Myers JL, Ryu JH. Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest. 2005;128(5):3310–5.
Churg A et al. Acute exacerbation (acute lung injury of unknown cause) in UIP and other forms of fibrotic interstitial pneumonias. Am J Surg Pathol. 2007;31(2):277–84.
Churg A, Wright JL, Tazelaar HD. Acute exacerbations of fibrotic interstitial lung disease. Histopathology. 2011;58(4):525–30.
Konishi K et al. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;180(2):167–75.
• Collard HR et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299(1):L3–7. This manuscript reports an investigation of the plasma biomarker profile for acute exacerbation of IPF, demonstrating it is predominantly suggestive of increased alveolar epithelial cell injury and/or activity.
Kakugawa T et al. Serum heat shock protein 47 levels are elevated in acute exacerbation of idiopathic pulmonary fibrosis. Cell Stress Chaperones. 2013.
Kahloon RA et al. Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am J Respir Crit Care Med. 2012;187(7):768–75.
Huie TJ et al. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: aetiology and outcomes. Respirology. 2010;15(6):909–17.
• Wootton SC et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(12):1698–702. This study demonstrated that viral infection is present in a minority of cases of acute exacerbation of IPF.
• Lee JS et al. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J. 2012;39(2):352–8. This study revealed that elevated pepsin levels are present in the bronchoalveolar lavage of patients with acute exacerbation of IPF, suggesting that aspiration is responsible for a subgroup of cases.
Tobin RW et al. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158(6):1804–8.
Raghu G et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J. 2006;27(1):136–42.
Raghu G et al. Sole treatment of acid gastroesophageal reflux in idiopathic pulmonary fibrosis: a case series. Chest. 2006;129(3):794–800.
Linden PA et al. Laparoscopic fundoplication in patients with end-stage lung disease awaiting transplantation. J Thorac Cardiovasc Surg. 2006;131(2):438–46.
• Lee JS et al. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: an analysis of data from three randomised controlled trials. Lancet Respir Med. 2013;1(5):369–76. This study revealed that anti-acid therapy may reduce the incidence of acute exacerbation of IPF in a clinical trial cohort.
Kondoh Y et al. Acute exacerbation of interstitial pneumonia following surgical lung biopsy. Respir Med. 2006;100(10):1753–9.
Sakamoto S et al. Acute exacerbation of idiopathic interstitial pneumonia following lung surgery in 3 of 68 consecutive patients: a retrospective study. Intern Med. 2011;50(2):77–85.
Sugiura H et al. Acute exacerbation of usual interstitial pneumonia after resection of lung cancer. Ann Thorac Surg. 2012;93(3):937–43.
Suzuki H et al. Risk of acute exacerbation of interstitial pneumonia after pulmonary resection for lung cancer in patients with idiopathic pulmonary fibrosis based on preoperative high-resolution computed tomography. Surg Today. 2011;41(7):914–21.
Yano M et al. Post-operative acute exacerbation of pulmonary fibrosis in lung cancer patients undergoing lung resection. Interact Cardiovasc Thorac Surg. 2012;14(2):146–50.
Mizuno Y et al. The importance of intraoperative fluid balance for the prevention of postoperative acute exacerbation of idiopathic pulmonary fibrosis after pulmonary resection for primary lung cancer. Eur J Cardiothorac Surg. 2012;41(6):e161–5.
Sakamoto K et al. Acute exacerbation of IPF following diagnostic bronchoalveolar lavage procedures. Respir Med. 2012;106(3):436–42.
Ghatol A, Ruhl AP, Danoff SK. Exacerbations in idiopathic pulmonary fibrosis triggered by pulmonary and nonpulmonary surgery: a case series and comprehensive review of the literature. Lung. 2012;190(4):373–80.
Rangappa P, Moran JL. Outcomes of patients admitted to the intensive care unit with idiopathic pulmonary fibrosis. Crit Care Resusc. 2009;11(2):102–9.
Al-Hameed FM, Sharma S. Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J. 2004;11(2):117–22.
Collard HR et al. Current diagnosis and management of idiopathic pulmonary fibrosis: a survey of academic physicians. Respir Med. 2007;101(9):2011–6.
Horita N et al. Tacrolimus and steroid treatment for acute exacerbation of idiopathic pulmonary fibrosis. Intern Med. 2011;50(3):189–95.
Abe S et al. Polymyxin B-immobilized fiber column (PMX) treatment for idiopathic pulmonary fibrosis with acute exacerbation: a multicenter retrospective analysis. Intern Med. 2012;51(12):1487–91.
Tachibana K et al. Polymyxin-B hemoperfusion for acute exacerbation of idiopathic pulmonary fibrosis: serum IL-7 as a prognostic marker. Sarcoidosis Vasc Diffuse Lung Dis. 2011;28(2):113–22.
Noma S et al. Two cases of acute exacerbation of interstitial pneumonia treated with polymyxin B-immobilized fiber column hemoperfusion treatment. Intern Med. 2007;46(17):1447–54.
Seo Y et al. Beneficial effect of polymyxin B-immobilized fiber column (PMX) hemoperfusion treatment on acute exacerbation of idiopathic pulmonary fibrosis. Intern Med. 2006;45(18):1033–8.
Oishi K et al. Association between cytokine removal by polymyxin B hemoperfusion and improved pulmonary oxygenation in patients with acute exacerbation of idiopathic pulmonary fibrosis. Cytokine. 2013;61(1):84–9.
Inase N et al. Cyclosporin A followed by the treatment of acute exacerbation of idiopathic pulmonary fibrosis with corticosteroid. Intern Med. 2003;42(7):565–70.
Sakamoto S et al. Cyclosporin A in the treatment of acute exacerbation of idiopathic pulmonary fibrosis. Intern Med. 2010;49(2):109–15.
Kondoh Y et al. Recombinant thrombomodulin in acute exacerbation of idiopathic pulmonary fibrosis, in B102. Interstitial lung disease: novel management and outcome strategies. American Thoracic Society. p. A3811.
Kondoh Y et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2010;27(2):103–10.
Moeller A et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(7):588–94.
Fujimoto K et al. Acute exacerbation of idiopathic pulmonary fibrosis: high-resolution CT scores predict mortality. Eur Radiol. 2012;22(1):83–92.
Compliance with ethics guidelines
Conflict of interest
Kerri Johannson declares that she has no conflicts of interest. Harold R. Collard is a consultant for Biogen, FibroGen, Genoa, Gilead, InterMune, MedImmune, Mesoblast, and Promedior. His institution receives money as a result. His institution also receives funding through grants he has from Boehringer–Ingelheim, Genentech, and NIH/NHLBI.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johannson, K.A., Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis: a proposal. Curr Respir Care Rep 2, 233–240 (2013). https://doi.org/10.1007/s13665-013-0065-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-013-0065-x