
Abstract
It has been proven that intra-articular injection of mesenchymal stromal cells (MSCs) can alleviate cartilage damage in osteoarthritis (OA) by differentiating into chondrocytes and protecting inherent cartilage. However, the mechanism by which the OA articular microenvironment affects MSCs’ therapeutic efficiency is yet to be fully elucidated. The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor involved in various cellular processes, such as osteogenesis and immune regulation. Tryptophan (Trp) metabolites, most of which are endogenous ligand for AHR, are abnormally increased in synovial fluid (SF) of OA and rheumatoid arthritis (RA) patients. In this study, the effects of kynurenine (KYN), one of the most important metabolites of Trp, were evaluated on the chondrogenic and chondroprotective effects of human umbilical cord-derived mesenchymal stromal cells (hUC-MSCs). hUC-MSCs were cultured in conditioned medium containing different proportions of OA/RA SF, or stimulated with KYN directly, and then, AHR activation, proliferation, and chondrogenesis of hUC-MSCs were measured. Moreover, the chondroprotective efficiency of short hairpin-AHR-UC-MSC (shAHR-UC-MSC) was determined in a rat surgical OA model (right hind joint). OA SF could activate AHR signaling in hUC-MSCs in a concentration-dependent manner and inhibit the chondrogenic differentiation and proliferation ability of hUC-MSCs. Similar results were observed in hUC-MSCs stimulated with KYN in vitro. Notably, shAHR-UC-MSC exhibited superior therapeutic efficiency in OA rat upon intra-articular injection. Taken together, this study indicates that OA articular microenvironment is not conducive to the therapeutic effect of hUC-MSCs, which is related to the activation of the AHR pathway by tryptophan metabolites, and thus impairs the chondrogenic and chondroprotective effects of hUC-MSCs. AHR might be a promising modification target for further improving the therapeutic efficacy of hUC-MSCs on treatment of cartilage-related diseases such as OA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is the most common degenerative disease of the joints worldwide, with the main pathological manifestations being joint deformity and articular cartilage destruction [1]. Traditional therapeutic strategies include surgery and medical therapy. Surgical options include articular lavage, microfracture, osteochondral allograft transfer system, and autologous chondrocyte implantation [2, 3], whereas medication treatments primarily include nonsteroidal anti-inflammatory drugs and injections of chondroprotective agents [4]. Although these interventions can alleviate OA to a certain extent and improve joint function, they do not promote articular cartilage regeneration or address the underlying causes of articular cartilage destruction [4].
Recently, with the development of cell tissue engineering, the use of stromal cells has become a new strategy and study hotspot for the treatment of various diseases. MSCs, a type of pluripotent stromal cell, are an ideal cell for the treatment of OA cartilage damage [5]. Many studies have shown that MSCs have great potential for promoting cartilage regeneration in animal models of OA, such as OA rats, OA rabbits, and OA dogs, among others. Preclinical studies and clinical trials have also shown that MSCs can be used to treat OA in the following ways: tissue engineering for MSCs transplantation, scaffold-free MSCs’ injection, and MSCs’ exosome injection [6,7,8]. Although the specific mechanism of MSCs in OA treatment and facilitation of cartilage repair is not yet clear, it may be related to the chondrogenic differentiation capacity and paracrine effects of MSCs. MSCs can differentiate into chondrocytes while recruiting resident progenitor chondrocytes to the cartilage site to promote cartilage matrix synthesis [9,10,11]. In addition, through paracrine effects, MSCs secrete a large number of cytokines and nutritional factors, exerting anti-inflammatory and nutritional effects on damaged cartilage [12]. Although MSCs have great advantages and potential in the treatment of OA and cartilage damage, MSC-related therapies are not yet routine clinical treatments [13]. In cell therapy, MSCs are susceptible to the influence of the local microenvironment [14]. Evidence suggests that the effects of MSC therapeutics are disturbed by the inflammatory environment. It has been reported that the inflammatory microenvironment of acute myocardial infarction inhibits the ability of stromal cells to promote myocardial regeneration. Some proinflammatory cytokines in the inflammatory microenvironment induce stromal cell death and hinder their proliferation and differentiation [15]. In addition, Lou et al. found that metabolic abnormalities and the inflammatory milieu affect the differentiation potential of adipose-derived mesenchymal stromal cells, impairing their function in maintaining tissue repair and homeostasis [16].
The homeostasis of the articular microenvironment in OA is dysregulated or even disordered. Therefore, it is necessary to study the influence of the articular microenvironment of patients with OA on the therapeutic efficiency of MSCs. Furthermore, studies have shown that the metabolic pathways for tryptophan are significantly upregulated with abnormally increased metabolites in the synovial fluid of OA patients [17, 18]. Tryptophan (Trp), an essential aromatic amino acid, is indispensable for protein synthesis. Most of the dietary Trp consumed is used for protein synthesis and the remainder is metabolized [19]. The metabolites of Trp, such as kynurenine (KYN), are generated by a series of endogenous enzymes or microbial metabolism, which enable them to bind and activate the aryl hydrocarbon receptor (AHR). This sequential process is termed the Trp–KYN–AHR pathway [20]. The activated Trp–KYN–AHR pathway regulates a variety of physiological and pathological processes, including growth, metabolism, emotions, and immunological responses [21]. AHR is a ligand-activated nuclear transcription factor found in a variety of tissues and cells; it belongs to the Per-ARNT-Sim homology domain family [22]. Upon ligand stimulation, AHR translocates from the cytoplasm to the nucleus to regulate the lower reaches of gene expression. Studies have shown that AHR is expressed in MSCs, where it participates in the regulation of the immune regulation of MSCs and affects migration function and the adipogenic and osteogenic differentiation potential of MSCs [23,24,25]. However, the effects of AHR on chondrogenic differentiation of MSCs remain unclear. Because the articular microenvironment is complicated in OA, MSCs are exposed to the synovial fluid (SF) after intra-articular injections. The levels of Trp and its metabolites, such as KYN, are reported to be abnormally increased in OA SF [17, 18]. KYN is an endogenous ligand of AHR. Thus, we speculated that the transactivation of AHR is induced by KYN and is involved in the inhibitory effects of OA SF on human umbilical cord mesenchymal stromal cells (hUC-MSCs). Whether the upregulated KYN pathway in SF can activate AHR and affect the chondrogenic and chondroprotective effects of MSCs has not yet been reported and was investigated in this study.
In this study, we investigated whether OA SF activates AHR and influences the proliferation and chondrogenic potential of hUC-MSCs. Among MSCs, hUC-MSCs have stronger proliferation and differentiation potential and are more appreciated in clinical practice [26]. In addition, hUC-MSCs have the characteristics of classic MSCs, proliferate rapidly, maintain their pluripotent differentiation ability after multiple passages, and have good cartilage regeneration potential [27].
In the present study, we found that OA SF could activate the AHR signaling pathway and inhibit chondrogenesis and proliferation of hUC-MSCs. Therefore, we investigated the role of the KYN–AHR axis in the inhibition of hUC-MSC chondrogenesis and proliferation. We used KYN to stimulate hUC-MSCs at different dosages in vitro, and similar results were obtained. However, this inhibition was blocked by AHR knockdown. Moreover, the complicated articular microenvironment has an unfavorable effect on the chondroprotective effects of hUC-MSCs. AHR knockdown in hUC-MSCs had a strong therapeutic effect in the OA rat model. These results clarified the impact of the KYN–AHR pathway on the chondrogenic and chondroprotective effects of hUC-MSCs, which provided a theoretical basis for the clinical application of MSCs in the treatment of cartilage destruction diseases such as OA.
Materials and methods
Synovial fluid collection
In this study, SF obtained from healthy individuals (who had undergone total knee replacement after knee trauma) was used as the control group, and SF obtained from OA and RA patients who had undergone therapeutic arthrocentesis was used as the OA SF group and the RA SF group, respectively. All SF samples were obtained from the First Affiliated Hospital of the Anhui Medical University. The study was conducted in accordance with the Declaration of Helsinki guidelines and approved by the research ethics board of the Anhui Medical University. All participants (or the relatives of those who were unable to sign) signed a written informed consent form before inclusion in the study. SF samples were transferred to heparin-treated tubes and centrifuged at 2000 rpm for 10 min at 4 °C to exclude cells and debris. Then, the supernatants were collected and filtered through a 0.2-μm pore size membrane to remove the remaining macromolecules and then stored at − 80 °C for future use.
Cells’ culture
hUC-MSCs (from an independent donor) were provided by Nanjing Kangya Biological Technology Co., Ltd. Cells were thawed at 37 °C for 1 min, then seeded into 25-cm2 culture flasks, and cultivated in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12, BI, Israel) containing 10% fetal bovine serum (FBS, BI, Israel) and 1% penicillin–streptomycin (Gibco). Cells were passaged at a split ratio of 1:3 when they reached 80% confluency. The medium was replaced every 3 days, and cells between the thirst and sixth passages were used in subsequent experiments.
Phenotype of hUC-MSCs
At the end of the third passage, the cells were collected and washed with phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA; Sigma-Aldrich, USA). The cells were then resuspended with 200 μL PBS before incubation with FITC/PE/APC-conjugated mouse anti-human antibodies (CD11b, CD34, CD45, CD73, CD90, or CD105) (BioLegend, USA) for 30 min in the dark at 4 °C. Fluorochrome and concentration-matched isotopes were used as controls (all IgG1, BioLegend, USA). Following two washes in PBS/2%FBS to remove unbound antibodies, cells were resuspended in 300 μL of PBS and analyzed using a flow cytometer (CytoFLEX, Beckman, USA).
Trilineage differentiation potential of hUC-MSCs
The multidirectional differentiation capabilities of hUC-MSCs were detected using commercial MSCgo™ differentiation kits (BI, Israel). hUC-MSCs were seeded in a 48-well plate at a density of 1 × 104 cells/well and cultured in adipogenic, osteogenic, or chondrogenic differentiation induction medium. The induction medium was changed every 3 days. On day 21, the cells were fixed and stained with Oil Red O for adipocytes, Alizarin red S for osteocytes, and Alcian blue for chondrocytes (Servicebio, China).
Synovial fluid treatment
To determine the effect of SF on chondrogenic differentiation of hUC-MSCs, cells were seeded at a density of 3 × 104 cells/well in 24-well plates in growth medium. At 80% confluence, cells were serum-starved overnight, and the medium was replaced with chondrogenic differentiation medium containing one of the following treatments: OA SF added to the medium at a ratio of 20% in the 20% OA SF group or OA SF added to the medium at a ratio of 40% in the 40% OA SF group. Similarly, the two RA SF groups also had the same proportions as the OA SF groups. The healthy SF group was used as a control. The medium was replaced every 3 days, and Alcian blue staining was performed 21 days after chondrogenic induction.
KYN treatment of hUC-MSCs
To determine the effect of KYN on the chondrogenic differentiation potential of UC-MSCs, hUC-MSCs were seeded in 24-well plates with growth medium. At approximately 80% confluence, the cells were serum-starved overnight using a chondrogenic differentiation medium containing 100 or 200 μM KYN (Sigma, USA) to replace the medium. The medium was replaced every 3 days, and Alcian blue staining was performed 21 days after chondrogenic induction.
Cell proliferation
The effect of SF and KYN on the activity of hUC-MSCs was measured using the Cell Counting Kit 8 assay (CCK-8, Beyotime, China). Cells were seeded in 96-well plates at an initial density of 1 × 103 cells/well, serum-starved overnight, and then cultured in DMEM/F12 containing 10% FBS and treated with SF or KYN for 7 days. At the indicated time points (days 1, 3, 5, or 7), cells were incubated with CCK-8 solution (10 μL/well) at 37 °C for 1 h. Each time point included six replicate wells. The optical density was determined at a wavelength of 450 nm using an enzyme-labeling instrument (BioTek Elx, Tecan, USA).
Lentiviral transfection
To establish stable AHR-low-expressing hUC-MSCs, a recombinant lentivirus containing green fluorescent protein (GFP) and stably expressing AHR shRNA (Gene Chem Co., Ltd., Shanghai, China) was constructed. A lentiviral vector containing GFP and a scrambled sequence served as a short hairpin negative control (shNC) (Gene Chem Co., Ltd., Shanghai, China). The shRNA sequences are as follows: shAHR, 5′-GCATAGAGACCGACTTAAT-3′ and shNC, 5′-TTCTCCGAACGTGTCACGT-3′. Lentivirus transfection was performed according to the manufacturer's instructions. hUC-MSCs were cultured in 12-well plates for 24 h and transduced with lentiviral particles using 8 μg/mL polybrene at a multiplicity of infection of 60. Antibiotic selection, which was conducted by adding puromycin (5 μg/mL, Sigma-Aldrich) and fluorescent cell sorting, was used to select stable knockdown cells.
RNA isolation and quantitative polymerase chain reaction (q-PCR)
Total RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific, USA) according to the manufacturer's instructions. Reverse transcription was performed using HiScript® II Q RT SuperMix for qRT-PCR (Vazyme Biotech, China) to synthesize cDNA. q-PCR was performed using the AceQ® qRT-PCR SYBR Green Master Mix (Vazyme Biotech, China) on a 7500 Real-Time PCR Detection System (Applied Biosystems, USA) with gene-specific primers. Primers were synthesized by Sangon Biotech (Shanghai, China). The sequences of primers used are as follows: AHR (forward 5′-CAGTGGTCCCAGCCTACAC-3′ and reverse 5-GACTGGCGTAGGTGATGTTG-3′), CYP1A1 (forward 5'-′CTCAGTACCTCAGCAGCCAC-3′ and reverse 5′-TTCTTCAGGCCTTTGGGGAC-3′), CYP1B1 (forward 5′-GACGCCTTTATCCTCTCTGCG-3′ and reverse 5′-ACGACCTGATCCAATTCTGCC-3′), SOX-9 (forward 5′-GGACTTCTGAACGAGAGCGAGA-3′ and reverse 5′-CGTTCTTCACCGACTTCCTCC-3′), COL2A1 (forward 5′-TGCATGAGGGCGCGGTA-3′ and reverse 5′-GGTCCTGGTTGCCGGACAT-3′), ACAN (forward 5′-ACATTGTGGGGCTAGAACGA-3′ and reverse 5′-CAGGAGGCTGCACAAGTTTT-3′), and GAPDH (forward 5′-ATGTTGCAACCGGGAAGGAA-3′ and reverse 5′-AGGAAAAGCATCACCCGGAG-3′). GAPDH was used as the reference gene. The relative mRNA expression levels in the groups were analyzed using the \(2^{{ - \Delta \Delta C_{{\text{T}}} }}\) method.
Western blot analysis
The cells were washed three times with PBS and lysed with RIPA lysis buffer. The cell lysates were centrifuged to obtain total cellular protein, and as for nuclear protein, nuclear fractions were extracted using the Nuclear Protein Extraction Kit (Sangon Biotech, China) according to the manufacturer's instructions. The protein concentration was determined using a BCA protein assay kit (BioChannel Biotechnology, China). Subsequently, the samples were denatured by boiling for 10 min in 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis buffer. For western blotting, the protein samples were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane (Millipore, USA). They were then incubated with the following primary antibodies: AHR (1:800, Santa Cruz, USA), SOX-9 (1:800, Santa Cruz, USA), COL2A1 (1:600, Proteintech, USA), Lamin B (1:500, Santa Cruz, USA), and anti-β-actin (1:1000; Cell Signaling Technology, USA). Membranes were then washed and treated with appropriate horseradish peroxidase-conjugated secondary antibodies and developed using electrochemiluminescence substrates (ELC, Thermo Fisher, USA). The relative band intensity was measured using the ImageJ software.
Immunofluorescence staining
Cells grown on glass coverslips were washed with ice-cold PBS, fixed with 4% formaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 5% bovine serum albumin. Then, the cells were incubated with appropriate primary antibodies at the following dilutions: anti-AHR (1:200, Santa Cruz, USA), anti-SOX-9 (1:200, Santa Cruz, USA), and anti-COL2A1 (1:150, Proteintech, USA) overnight at 4 °C. The following day, cells were treated with the following fluorescence-labeled secondary antibodies: anti-mouse IgG 488 (1:200, Biologend, USA), anti-goat IgG 594 (1:200, Biologend, USA), and anti-mouse IgG 647 (1:200, Biologend, USA) for 2 h at room temperature in the dark. After washing with PBS, the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma, USA) for 10 min, and an anti-fluorescence quencher was added. Images were obtained using a confocal microscope (Leica TCS SP8, Germany).
Alcian blue staining
hUC-MSCs were cultured in a chondrogenic differentiation medium to induce chondrogenic differentiation. On day 21 after chondrogenic induction, the cells were washed twice with PBS, fixed for 30 min with 4% formaldehyde solution, and stained overnight with Alcian blue (1% acetic acid, pH 2.5). Then, the cells were washed three times with 0.1 N HCL and imaged using a microscope. In addition, the total sulfated GAG content in chondrocyte spheroids was quantified. Briefly, chondrocyte spheroids were treated with 8 M guanidine HCL containing 0.05 M acetate (pH 5.8) and proteinase inhibitors overnight. The absorbance of the eluent was measured at 600 nm using a microplate reader as previously described [15].
Animal studies
All animal protocols were performed in accordance with laboratory animal care and use guidelines and were approved by the Animal Ethics Committee of the Institute of Clinical Pharmacology Laboratory at Anhui Medical University.
Induction of osteoarthritis model
Thirty-two male Sprague–Dawley (SD) rats (approximately 12 weeks old) were purchased from SPF Biotechnology Co. (Beijing, China) and housed in a specific pathogen-free (SPF) animal laboratory with a 12:12 h light/dark cycle and controlled temperature (23–25 °C). After 1 week of acclimatization, according to previous research [28], the OA rat model was established by completely transecting the medial collateral ligament and medial meniscus, removing the meniscus, and cutting off the anterior cruciate ligament without damaging the tibial surface (OA-like joint discomfort in the right hind joint). After surgery, all rats received an intramuscular injection of penicillin sodium (10 mg/kg) in the right medial thigh for 3 days to prevent infection. The rats were then randomly divided into the following four groups (n = 8 per group): (1) sham group: saline, 100 μL, (2) OA group: saline, 100 μL, (3) OA + shNC-hUC-MSCs group: 5 × 107 shNC-hUC-MSCs cells/100 mL saline, and (4) OA + shAHR-hUC-MSCs group: 5 × 107 shAHR-hUC-MSCs cells/100 mL saline. Four weeks after injection, the rats were sacrificed, and knee samples were harvested for X-ray examination, overall scoring, and pathological and histological tests.
Establishment of adjuvant-induced arthritis model
To induce arthritis, animals were inoculated by subcutaneous injection of 0.1 mL/rat CFA solution (Sigma Chemical Co., USA) into the footpad of the right hind paw for 2 consecutive days. Each 1 mL of CFA contained 1 mg of heat-killed and dried mycobacterium tuberculosis, 0.85 mL of paraffin oil, and 0.15 mL of mannite monooleate.
Radiography
Before the animals were euthanized, an X-ray examination was performed on knee joint samples to observe osteophyte formation, joint space, and cartilage damage. All rats were anesthetized with ether at the end of the protocol. The rats were placed in a supine position, and radiographs of the right knee were taken. Two independent investigators blindly evaluated the radiographic grading according to the Kellgren–Lawrence scoring system. The Kellgren and Lawrence system provides a score of severity from 0 to 4 as follows: grade 0, no radiographic features of OA are present; grade 1, doubtful joint space narrowing (JSN) and possible osteophytic lipping; grade 2, definite osteophytes and possible JSN on anteroposterior weight-bearing radiograph; grade 3, multiple osteophytes, definite JSN, sclerosis, possible bony deformity; grade 4, large osteophytes, marked JSN, severe sclerosis, and definite bony deformity [29].
Histology and immunohistochemical analysis
After the knee joints were opened and disarticulated, the gross morphological lesions on the rat tibial plateaus were visualized, and cartilage lesions and fibrillation were quantified according to the Osteoarthritis Research Society International (OARSI) guidelines [28]. After collecting knee joint samples and removing the tibiofemoral joints, the remaining femoral condyles were fixed in neutral buffered formalin (containing 4% formaldehyde) for 24 h. The fixed femoral condyles were decalcified in EDTA for 3 weeks and embedded in paraffin. Tissues were embedded in paraffin and sectioned into 5-μm-thick sections. Serial sections were obtained from the medial and lateral compartments at 200-μm intervals. The selected sections were deparaffinized in xylene and rehydrated through a graded series of ethanol washes, followed by safranin-O/fast green staining (Servicebio, China). Cartilage degeneration was assessed using Mankin’s score [30]. The OARSI and Mankin scoring were performed by three independent blinded observers.
For immunohistochemistry, deparaffinized sections were soaked in EDTA (pH 9.0) for antigen retrieval using microwave-assisted irradiation. The sections were placed in 3% hydrogen peroxide solution and incubated at room temperature for 25 min in the dark, followed by blocking with 5% BSA at room temperature for 30 min. Then, the sections were incubated with the primary antibodies, collagen II (Abcam, UK), and aggrecan (Abcam, UK) at 4 °C overnight, followed by incubation with secondary antibodies (Abcam, UK) at room temperature for 60 min the next day. After thorough washing, 3,3′-diaminobenzidine (DAB)-peroxidase substrate and hematoxylin solution (Servicebio, China) were added.
KYN determination in synovial fluid by high-performance liquid chromatography (HPLC)
KYN concentrations were measured by HPLC as previously described [31]. Briefly, frozen samples were thawed immediately prior to the investigation. Proteins were precipitated with trichloroacetic acid, mixed, and centrifuged at 12,000 rpm and 4 °C. For the measurement, 10 μL of clear supernatant was injected into the HPLC system using Agilent TC-C18 columns (250 mm length, 5 μm grain size) for separation. Kynurenine was detected at a wavelength of 360 nm using a fluorescence detector (Agilent, G1315D). Data were recorded using the Agilent ChemStation software.
Statistical analysis
Statistical analysis was performed using SPSS v. 25.0, and multi-factor analysis of variance (ANOVA) was applied for comparison among multiple groups. All experiments were repeated in three independent batches and the results are presented as mean ± SEM; P < 0.05 was considered statistically significant. Cell culture samples and animals were randomly assigned to each group, with approximately equivalent numbers in each group, and all samples were analyzed in a blinded manner.
Results
hUC-MSCs’ identification and its characteristics
Frozen hUC-MSCs were resuscitated and expanded under standard conditions. After 6 h of resuscitation, the cells showed adherence and rapid proliferation. After 48 h of culture, the cells reached 80% confluence with fibroblast-like morphology and spindle-shaped appearance. There was no discernible variation in morphology or plastic adhesion characteristics until passage 5 (Fig. 1A).

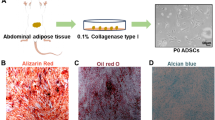
Identification of hUC-MSCs. A Growth and morphology of hUC-MSCs at first passages (P1) and fifth passages (P5) respectively (scale bar = 500 μm). B Cell proliferation of P1 and P5 was assessed by the CCK-8 assay (n = 3). C The markers for hUC-MSCs were detected by flow cytometry and hUC-MSCs were positive for CD73, CD90, and CD105, while negative for CD11b, CD34, and CD45 (n = 3, all histograms show specific membrane antigen in green and control isotype-specific IgGs in red). D hUC-MSCs differentiation potentials. Adipogenic differentiation capacity (Oil Red O staining), osteogenic differentiation capacity (Alizarin Red staining), and chondrogenic differentiation capacity (Alcian blue staining) (scale bar = 250 μm) (n = 3)
CCK-8 was used to detect the variation in the proliferation of hUC-MSCs from passages 1 and 5. The results showed no significant difference in the growth of hUC-MSCs after 5 passages (Fig. 1B).
Flow cytometry was used to identify hUC-MSC phenotypes. hUC-MSCs were positive for the canonical MSC markers CD73 (99.37%), CD90 (99.16%), and CD105 (99.19%), and negative for the hematopoietic markers CD11b (0.49%), CD34 (0.36%), and CD45 (0.34%) (Fig. 1C), indicating that hUC-MSCs express most of the consensus MSC markers, suggesting that these cells possess MSC-like characteristics. The differentiation potential of hUC-MSCs was also examined. As shown in Fig. 1D, Oil Red O, Alizarin red S, and Alcian blue staining revealed the adipogenic, osteogenic, and chondrogenic differentiation abilities of hUC-MSCs, respectively.
RA SF and OA SF activated AHR and inhibited the chondrogenic and proliferative capacities of hUC-MSCs
To explore the effect of SF from patients with OA or RA on the chondrogenesis of hUC-MSCs, cells were cultured in chondrogenic differentiation medium supplemented with OA or RA SF at an indicated concentration of 20% or 40% for 14 days. OA or RA SF stimulation was markedly upregulated, in a concentration-dependent manner, the mRNA expression of AHR and its downstream target genes, including CYP1A1 and CYP1B1 (Fig. 2A), whereas significantly decreased the expression levels of SOX-9, COL2A1, and ACAN (Fig. 2B). Western blotting results showed that OA or RA SF downregulates the expression of SOX-9 and COL2A1 (Fig. 2C, D), and promotes the nuclear translocation of AHR (Fig. 2E, F). In addition, fluorescence confocal assays revealed a similar trend (Fig. 2G–J).

OA/RA SF activates AHR and inhibits the chondrogenesis and proliferation of hUC-MSCs. A Relative mRNA expression of AHR, and downstream genes CYP1A1 and CYP1B1 and B cartilage matrix genes SOX-9, COL2A1, and ACAN at day 14 of chondrogenic differentiation (n = 5). C–F Protein levels of AHR, COL2A1, and SOX9 were analyzed using western blotting at day 14 of chondrogenic differentiation. Lamin-B1 and β-actin served as the loading controls, respectively. The data represents two independent experiments. (n = 5). G–J Immunofluorescence staining of AHR, COL2A1 (G, H), and SOX-9 (I, J), and the mean fluorescence intensity was quantified (scale bar = 25 μm, scale bar = 20 μm) (n = 5). K, L Alcian Blue staining for evaluating chondrogenic differentiation of hUC-MSCs and the staining eluent was quantified (scale bar = 200 μm) (n = 5). M The CCK-8 assay was used to detect the cell vitality of UC-MSCs treated with OA or RA SF for 1, 3, 5, and 7 days (n = 4). Each experiment was repeated four-to-five times independently, and the data were reported as mean ± SEM. *P < 0.05, **P < 0.01
Similarly, extracellular matrix (ECM) secretion also decreased in the SF-stimulated groups, as determined by Alcian blue staining and quantification on day 14 (Fig. 2K, L).
Next, we examined the effect of SF on hUC-MSC proliferation using the CCK-8 assay. As shown in Fig. 2M, treatment of hUC-MSCs with OA SF or RA SF for 3, 5, and 7 days inhibited their growth compared to the control group. These results suggest that OA SF and RA SF activated AHR and inhibited chondrogenesis and proliferation in hUC-MSCs.
KYN treatment activated AHR and suppressed chondrogenesis and proliferation of hUC-MSCs
KYN, a tryptophan metabolite found in the synovial fluid of patients with OA and RA, is an endogenous ligand for AHR. Thus, we speculated that the activation of AHR in hUC-MSCs by SF may be due to the effect of KYN. We detected KYN content in SF from OA patients, RA patients, and normal donors by HPLC. We found that KYN was expressed at much higher levels in the SF of OA and RA patients than in normal donors, and KYN concentration in OA SF was about 20 µM, which was higher than that in RA SF (Fig. 3A). Therefore, we first tested whether pathological level of KYN (20 µM) could affect the chondrogenesis of hUC-MSCs. The results showed that 20 µM KYN downregulated the protein expression of COL2A1 and SOX-9 (Supplementary Fig. 1A and 1C), and the mRNA level of COL2A1, SOX-9, and ACAN in hUC-MSCs (Supplementary Fig. 1F). Meanwhile, KYN (20 µM) promoted nuclear AHR expression (Supplementary Fig. 1B and 1D) and AHR-regulated CYP1A1 and CYP1B1 mRNA levels (Supplementary Fig. 1E), which indicated the activation of AHR in hUC-MSCs. Next, we stimulated hUC-MSCs with KYN (100 µM and 200 µM) and examined the nuclear translocation of AHR. KYN treatment significantly increased the nuclear translocation of AHR, indicating that KYN can activate AHR signaling (Fig. 3E, F). This result was further supported by the increased mRNA expression of AHR-regulated metabolic enzymes, including CYP1A1 and CYP1B1 (Fig. 3B). Moreover, to clarify whether AHR activation regulates chondrogenesis of hUC-MSCs, the levels of chondrogenic differentiation-related SOX-9 and COL2A1 mRNA were determined. q-PCR results revealed that KYN significantly increased the expression of AHR, CYP1A1, and CYP1B1 mRNA in a dose-dependent manner (Fig. 3B), and suppressed the mRNA expression levels of SOX-9, COL2A1, and ACAN (Fig. 3C). Similarly, western blotting and immunofluorescence showed that KYN promotes the nuclear translocation of AHR (Fig. 3E, F); however, that of SOX-9 and COL2A1 was markedly reduced by KYN (Fig. 3F, G). Additionally, as shown in Fig. 3H, the ECM secretion of hUC-MSCs in the KYN treatment groups was suppressed compared to that in the control group.

KYN–AHR axis activation suppressed the chondrogenic and proliferative activity of hUC-MSCs. A The content of KYN in SF from OA, RA patients, and normal donors by HPLC. KYN was expressed at a much higher level in synovial fluid of OA and RA patients than normal donors (n = 3–5). For 14 days, hUC-MSCs were incubated with KYN (0, 100, or 200 M) in the presence of chondrogenic media. q-PCR assay was used to detect the mRNA expression level of AHR and its downstream target genes CYP1A, CYP1B1 (B) and chondrogenic markers SOX-9, COL2A1, and ACAN (C) (n = 5). D, E The protein expressions of AHR, SOX-9, and COL2A1 were detected by Western blot. Lamin-B1 and β-actin served as the loading controls, respectively. The data represent two independent experiments (n = 5). F, G Immunofluorescence staining of AHR, COL2A1, and SOX-9. At day 14 of chondrogenic differentiation, and the mean fluorescence intensity was quantified (scale bar = 25 μm, scale bar = 20 μm) (n = 5). H Alcian blue staining and relative quantitative analysis were used to evaluate the effects of KYN on the chondrogenic differentiation of hUC-MSCs (scale bar = 200 μm) (n = 5). I The CCK-8 assay was used to detect the cell vitality of hUC-MSCs treated with KYN for 1, 3, 5, and 7 days (n = 4). Each experiment was repeated four-to-five times independently, and data were reported as mean ± SEM. *P < 0.05, **P < 0.01
CCK-8 assays were used to explore the effects of AHR activation on the proliferation of hUC-MSCs. There was no difference in cell viability among the three groups on day 1; however, on days 3, 5, and 7, the proliferation of hUC-MSCs in the KYN treatment groups was reduced compared with the control group (Fig. 3I).
Establishment of a short hairpin AHR-UC-MSC (shAHR-hUC-MSC) stable cell line
The effects of AHR on chondrogenesis and proliferation were evaluated by gene knockdown experiments using shRNA. Following puromycin selection, the knockdown efficiency was verified by q-PCR, western blotting, and fluorescence staining. q-PCR and western blot results revealed that the gene and protein expression levels of AHR in the shAHR group were significantly downregulated compared to those in the shNC group (Fig. 4A–C). Fluorescent staining revealed that the transfection efficiency was > 90% (Fig. 4D). To verify whether the lentivirus changed the phenotype of hUC-MSCs, flow cytometry was used to analyze the surface markers of shAHR-hUC-MSCs, and shAHR-hUC-MSCs were confirmed to be positive for the MSC markers CD73 (98.13%), CD90 (98.43%), and CD105 (98.79%) and negative for CD11b (0.26%), CD34 (0.69%), and CD45 (0.96%) (Fig. 4E).

Lentiviral transfection to knockdown AHR in hUC-MSCs. A Relative mRNA expression of AHR in control, shNC, and shAHR groups (n = 5). B, C Protein levels of AHR in the control, shNC, and shAHR groups (n = 5). D Images of GFP-positive hUC-MSCs under a normal microscope and a fluorescence microscope (scale bar = 200 μm). E The phenotype identification of shAHR-hUC-MSCs using flow cytometry (n = 3, all histograms show specific membrane antigen in green and control isotype-specific IgGs in red). The data are shown as mean ± SEM for triplicate. *P < 0.05
AHR knockdown prevented KYN from inhibiting chondrogenesis and proliferation of hUC-MSCs
We induced chondrogenic differentiation of hUC-MSCs, shNC-hUC-MSCs, and shAHR-hUC-MSCs in the presence of 200 μM KYN. As shown in Fig. 5A and B, compared with the control group, KYN treatment significantly induced AHR, CYP1A1, and CYP1B1 mRNA expression, but decreased SOX-9, COL2A1, and ACAN expression in KYN and KYN + shNC-hUC-MSCs. AHR knockdown significantly attenuated KYN-induced AHR expression and restored SOX-9 and COL2A1 levels. A similar trend of protein expression was revealed by western blotting and immunofluorescence staining, AHR knockdown remarkedly inhibited the activation of AHR (Fig. 5D, E) and upregulated SOX-9 and COL2A1 expression compared with shNC group (Fig. 5D, F).

AHR knockdown reversed the inhibition effect of KYN treatment on chondrogenesis and proliferation of hUC-MSCs. A, B AHR, CYP1A, CYP1B, SOX-9, COL2A1, and ACAN gene expression were evaluated by quantitative q-PCR (n = 5). C, D Western blot analysis of AHR, SOX-9, and COL2A1 protein level. Lamin-B1 and β-actin served as the loading controls respectively. The data represent two independent experiments. (n = 5). E, F Immunofluorescent staining of AHR, COL2A1, and SOX-9 in hUC-MSCs, and the mean fluorescence intensity was quantified (scale bar = 25 μm, scale bar = 20 μm) (n = 5). G Alcian blue staining and quantification (scale bar = 200 μm) (n = 5). H The CCK-8 assay was used to detect the cell vitality of shAHR-hUC-MSCs treated with KYN for 1, 3, 5, and 7 days (n = 4). Each experiment was repeated four-to-five times independently, and data were reported as mean ± SEM. *P < 0.05, **P < 0.01
Following AHR knockdown, the decreased ECM secretion of hUC-MSCs induced by KYN treatment was effectively abrogated (Fig. 5G). Additionally, the decreased cell viability of hUC-MSCs caused by KYN treatment was also restored after AHR knockdown (Fig. 5H).
shAHR-hUC-MSCs showed a stronger chondroprotective effect on OA rats
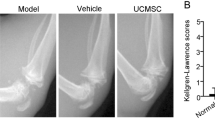
The homeostasis of the articular microenvironment in OA is dysregulated or even disordered. It is unclear whether the cartilage protective efficacy of hUC-MSCs is affected after injection into the joint cavity. Our results suggest that OA SF affects chondrogenesis and proliferation of hUC-MSCs, probably by activating the KYN–AHR axis in vivo. To investigate the role of AHR in the repair of cartilage damage by hUC-MSCs in vivo, a surgically induced OA rat model was established. All OA rat models received different treatments as described in the Methods section. X-rays were used to detect knee joint lesions. The imaging results showed that, compared with the sham group, the wear and tear of the cartilage surface was significantly more in the OA group, and the shNC group also showed mild symptoms of joint wear (Fig. 6A). The cartilage surface was smoother in both the shAHR and sham groups, and the joint space was more obvious than that in the OA and shNC groups (Fig. 6A). The radiographic grading score system suggested that cartilage lesions in the OA and shNC-hUC-MSC groups were significantly higher than those in the shAHR group (Fig. 6A). The gross morphology of the knee cartilage and the OARSI score showed that compared with the shNC group, the shAHR group could significantly attenuate cartilage damage and had a better therapeutic effect on cartilage regeneration (Fig. 6B). Furthermore, safranin-O and fast green staining revealed that both the shNC-hUC-MSC and shAHR-hUC-MSC groups attenuated cartilage lesions and promoted cartilage regeneration, with the shAHR group having more cellularity and fewer superficial layer losses than the shNC group (Fig. 6C). Immunohistochemistry revealed that cartilage matrix collagen II (Fig. 6D) and aggrecan (Fig. 6E) expression levels in the shAHR group were more substantial than those in the shNC group and slightly lower than those in the sham group. These findings show that shAHR-hUC-MSCs significantly reduced cartilage damage in the rat model and provided better cartilage protection than shNC-hUC-MSCs in the rat OA model.

The effect of shAHR-hUC-MSCs on cartilage protection in vivo. A X-ray was used to evaluate the severity of OA in the knee joint before the animals were euthanized and the degree of cartilage lesions and osteophyte formation were assessed by the Kellgren–Lawrence scoring system (n = 5). B Gross morphology of articular cartilage damage of rats from different group, and cartilage destruction was evaluated using the OARSI scoring system (n = 5). C Safranin O-fast green staining for histopathologically observing the articular cartilage damage of rats from different group, and Mankin’s score was used to evaluate the degree of cartilage destruction (scale bar = 200 μm, scale bar = 100 μm) (n = 5). D, E Immunohistochemical staining was used for assessing the expression of Collagen II and Aggrecan, and their quantitative analysis (scale bar = 200 μm, scale bar = 100 μm) (n = 5). F, G Immunohistochemical staining was used for assessing the expression of IDO1 and TDO2 in the synovial tissues of healthy (post-trauma) people, OA, and RA patients (scale bar = 200 μm, scale bar = 100 μm) (n = 3). H, I Immunohistochemical staining was used for assessing the expression of IDO1 and TDO2 in the synovial tissues of normal, OA, and RA rats (scale bar = 200 μm, scale bar = 100 μm) (n = 3). Data were expressed as means ± SEM. *P < 0.05, **P < 0.01
In addition, we detected the expression of indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase 2 (TDO2), both of which are rate-limiting enzymes in the TRP–KYN metabolic pathway, in the synovial tissues of normal people (post-trauma), OA patients, and RA patients, as well as in the synovial tissues of normal rats, OA rats, and adjuvant arthritis (AA, animal model for RA) rats. The expression of IDO1 and TDO2 was significantly higher in the synovial tissues of patients with OA and RA than in those of healthy controls (Fig. 6F, G), and the same results were observed in the synovial tissues of animal models (Fig. 6H, I). These findings suggest that the TRP–KYN metabolic pathway is abnormally activated in the joints of OA and RA patients and that the resulting alteration of the articular cavity microenvironment may be detrimental to the therapeutic effects of MSC articular injection therapy.
Discussion
MSCs, especially UC-MSCs, have been proved to be a prospective candidate for management of cartilage-related diseases both in preclinical and clinical studies due to their anti-inflammation, immune privilege, and multilineage differentiation abilities [6, 32]. However, despite aforesaid superiorities, as a cell-based therapeutic strategy, MSCs were sensitive to environmental issues. Pathological microenvironment, such as inflammation and oxidative stress, altered the therapeutic potential of MSCs via inducing apoptosis/autophagy and transforming paracrine signaling [15, 33, 34]. In this study, we demonstrated that the chondrogenic efficiency of hUC-MSCs is influenced by the articular microenvironment in which they are present. The SF of OA patients suppresses chondrogenic differentiation and proliferation of hUC-MSCs through ligand-mediated transactivation of AHR. Furthermore, AHR knockdown blocked the preceding inhibition effect and improved the therapeutic efficiency of hUC-MSCs for cartilage regeneration in an OA rat model.
The therapeutic ability of MSCs in cartilage damage has been shown to depend upon their chondrogenic potential and paracrine effects. MSCs can differentiate into chondrocytes, induce the differentiation of resident progenitor cells, and secrete large levels of cytokines to promote cartilage regeneration [35, 36]. However, chondrogenic differentiation of MSCs in vivo is unstable and susceptible to the microenvironment [15, 33]. Studies have shown that catabolic factors, such as IL-1 or TNF-α in OA SF, inhibit chondrogenesis of MSCs; however, blocking IL-1 or TNF-α only partially overcomes the inhibitory effect [37], indicating that there are additional factors present in OA SF that participate in the inhibition of the chondrogenic differentiation of MSCs. In our study, we verified that OA SF inhibited chondrogenesis and impaired the proliferation of hUC-MSCs. RA SF, as the control, also had similar inhibitory effects on hUC-MSCs, but compared with OA SF groups, RA SF groups had stronger suppression effects, which may be due to the higher levels of proinflammatory cytokines compared with OA SF [38, 39]. Interestingly, we also found that AHR in hUC-MSCs was activated after OA SF or RA SF treatment in a concentration-dependent manner.
AHR, a ligand-activated nuclear transcription factor that exists in MSCs, functions as a master regulator of cell signaling pathways and participates in cell proliferation, differentiation, and apoptosis [24]. On stimulation, AHR translocates from the cytoplasm to the nucleus to mediate downstream signal transduction. Several studies observed the regulatory function of AHR on the plasticity of MSCs. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-activated AHR has been shown to impair the osteogenesis of MSCs via inhibiting a variety of downstream osteogenic-related signals [40, 41]. Meanwhile, benzo(a)pyrene-activated AHR inhibit adipogenesis of human bone marrow-derived MSCs in an AHR-dependent manner [42]. These results suggest a role for AHR in modulating MSCs’ differentiation, whereas the functions of AHR on MSCs’ chondrogenesis are remain unclear. The levels of tryptophan and its metabolites, such as KYN, are abnormally increased in OA SF [17, 18]. KYN is an endogenous ligand of AHR. Thus, we speculated that transactivation of AHR was induced by KYN and was involved in the inhibitory effects of OA SF on hUC-MSCs. To verify this, hUC-MSCs were directly incubated with KYN at different doses, and KYN induced AHR activation and suppressed chondrogenesis and proliferation of hUC-MSCs.
To further confirm the role of AHR in chondrogenic differentiation and proliferation of hUC-MSCs, we used lentivirus transfection to knock down AHR expression to obtain shAHR-hUC-MSCs, which retained the characteristics of MSCs. KYN significantly rescued chondrogenesis and proliferation of shAHR-hUC-MSCs compared with the hUC-MSCs group. These results indicate that AHR impedes chondrogenic differentiation and proliferation of hUC-MSCs. In addition to the in vitro study, an OA rat model based on surgery-induced knee joint instability, which can precisely simulate the symptoms and pathological manifestations of human OA, was used to evaluate the effect of shAHR-hUC-MSCs in vivo. The intra-articular injection of shAHR-hUC-MSCs significantly attenuated cartilage damage caused by OA and had a better effect on cartilage regeneration than shNC-hUC-MSCs. Based on our findings, the elevated KYN levels in OA SF and subsequent activation of AHR are highly likely part of the reason for the impairment of chondrogenesis and chondroprotective effect of hUC-MSCs. It is unclear, however, how the KYN levels changed during OA progression, and whether other TRP metabolites, such as kynurenic acid and quinolinic acid, exert similar inhibitory effect on chondrogenesis of MSCs as KYN, as they are endogenous ligands for AHR, as well [43].
Conclusion
In this study, we demonstrated that the OA articular microenvironment is not conducive to the therapeutic effect of hUC-MSCs and is related to the activation of the AHR pathway by tryptophan metabolites, which impairs the chondrogenic and chondroprotective effects of hUC-MSCs. AHR may be a promising target for further improving the therapeutic efficacy of hUC-MSCs in the treatment of cartilage-related diseases such as OA (Fig. 7). In addition, the efficiency of Trp metabolic enzymes’ (TDO2 and IDO1) inhibitors on improving the MSCs’ chondroprotective function in OA needs to be tested in further study, as they may inhibit AHR activation via block KYN production. All in all, our findings may help develop more effective cartilage regeneration strategies for OA.

Activation of KYN–AHR axis impairs the chondrogenesis and chondroprotective effects of hUC-MSCs. KYN–AHR pathway on the chondrogenic and chondroprotective effects of hUC-MSCs, which provided a theoretical basis for the clinical MSCs’ applications in the treatment of cartilage destruction diseases, such as OA
References
Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet (London, England). 2019;393(10182):1745–59.
Schmidt I. Surgical treatment options in thumb carpometacarpal osteoarthritis: a recent literature overview searching for practice pattern with special focus on total joint replacement. Curr Rheumatol Rev. 2015;11(1):39–46.
Hulme CH, Wilson EL, Peffers MJ, Roberts S, Simpson DM, Richardson JB, Gallacher P, Wright KT. Autologous chondrocyte implantation-derived synovial fluids display distinct responder and non-responder proteomic profiles. Arthritis Res Ther. 2017;19(1):150.
Hermann W, Lambova S, Muller-Ladner U. Current treatment options for osteoarthritis. Curr Rheumatol Rev. 2018;14(2):108–16.
Malekpour K, Hazrati A, Zahar M, Markov A, Zekiy AO, Navashenaq JG, Roshangar L, Ahmadi M. The potential use of mesenchymal stem cells and their derived exosomes for orthopedic diseases treatment. Stem Cell Rev Rep. 2022;18(3):933–51.
Lee W-S, Kim HJ, Kim K-I, Kim GB, Jin W. Intra-articular injection of autologous adipose tissue-derived mesenchymal stem cells for the treatment of knee osteoarthritis: a phase IIb, randomized, placebo-controlled clinical trial. Stem Cells Transl Med. 2019;8(6):504–11.
Wu J, Kuang L, Chen C, Yang J, Zeng W-N, Li T, Chen H, Huang S, Fu Z, Li JJB. miR-100-5p-abundant exosomes derived from infrapatellar fat pad MSCs protect articular cartilage and ameliorate gait abnormalities via inhibition of mTOR in osteoarthritis. Biomaterials. 2019;206:87–100.
Chen X, Shi Y, Xue P, Ma X, Li J, Zhang J. Mesenchymal stem cell-derived exosomal microRNA-136-5p inhibits chondrocyte degeneration in traumatic osteoarthritis by targeting ELF3. Arthritis Res Ther. 2020;22(1):1–13.
Yang Y, Lin H, Shen H, Wang B, Lei G, Tuan RS. Mesenchymal stem cell-derived extracellular matrix enhances chondrogenic phenotype of and cartilage formation by encapsulated chondrocytes in vitro and in vivo. Acta Biomater. 2018;69:71–82.
Diekman BO, Guilak F. Stem cell-based therapies for osteoarthritis: challenges and opportunities. Curr Opin Rheumatol. 2013;25(1):119–26.
Kangari P, Talaei-Khozani T, Razeghian-Jahromi I, Razmkhah M. Mesenchymal stem cells: amazing remedies for bone and cartilage defects. Stem Cell Res Ther. 2020;11(1):492.
Cosenza S, Ruiz M, Toupet K, Jorgensen C, Noel D. Mesenchymal stem cells derived exosomes and microparticles protect cartilage and bone from degradation in osteoarthritis. Sci Rep. 2017;7(1):16214.
Li N, Gao J, Mi L, Zhang G, Zhang L, Zhang N, Huo R, Hu J, Xu K. Synovial membrane mesenchymal stem cells: past life, current situation, and application in bone and joint diseases. Stem Cell Res Ther. 2020;11(1):381.
Lin S, Lee WYW, Feng Q, Xu L, Wang B, Man GCW, Chen Y, Jiang X, Bian L, Cui L, et al. Synergistic effects on mesenchymal stem cell-based cartilage regeneration by chondrogenic preconditioning and mechanical stimulation. Stem Cell Res Ther. 2017;8(1):221.
Zhao Y, Yang X, Li S, Zhang B, Li S, Wang X, Wang Y, Jia C, Chang Y, Wei W. sTNFRII-Fc modification protects human UC-MSCs against apoptosis/autophagy induced by TNF-alpha and enhances their efficacy in alleviating inflammatory arthritis. Stem Cell Res Ther. 2021;12(1):535.
Louwen F, Ritter A, Kreis N, Yuan JJOR. Insight into the development of obesity: functional alterations of adipose-derived mesenchymal stem cells. Obes Rev. 2018;19(7):888–904.
Nowicka-Stazka P, Langner E, Turski W, Rzeski W, Parada-Turska J. Quinaldic acid in synovial fluid of patients with rheumatoid arthritis and osteoarthritis and its effect on synoviocytes in vitro. Pharmacol Rep. 2018;70(2):277–83.
Kang KY, Lee SH, Jung SM, Park SH, Jung BH, Ju JH. Downregulation of tryptophan-related metabolomic profile in rheumatoid arthritis synovial fluid. J Rheumatol. 2015;42(11):2003–11.
Song P, Ramprasath T, Wang H, Zou MH. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell Mol Life Sci. 2017;74(16):2899–916.
Sun M, Ma N, He T, Johnston LJ, Ma X. Tryptophan (Trp) modulates gut homeostasis via aryl hydrocarbon receptor (AhR). Crit Rev Food Sci Nutr. 2020;60(10):1760–8.
Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan-kynurenine-aryl hydrocarbon axis. Clin Cancer Res. 2019;25(5):1462–71.
Shinde R, McGaha TL. The aryl hydrocarbon receptor: connecting immunity to the microenvironment. Trends Immunol. 2018;39(12):1005–20.
Heo JS, Lim JY, Pyo S, Yoon DW, Lee D, Ren WX, Lee SG, Kim GJ, Kim J. Environmental benzopyrene attenuates stemness of placenta-derived mesenchymal stem cells via aryl hydrocarbon receptor. Stem Cells Int. 2019;2019:7414015.
Xu T, Zhou Y, Qiu L, Do DC, Zhao Y, Cui Z, Wang H, Liu X, Saradna A, Cao X, et al. Aryl hydrocarbon receptor protects lungs from cockroach allergen-induced inflammation by modulating mesenchymal stem cells. J Immunol. 2015;195(12):5539–50.
Cui Z, Feng Y, Li D, Li T, Gao P, Xu T. Activation of aryl hydrocarbon receptor (AhR) in mesenchymal stem cells modulates macrophage polarization in asthma. J Immunotoxicol. 2020;17(1):21–30.
Xie Q, Liu R, Jiang J, Peng J, Yang C, Zhang W, Wang S, Song J. What is the impact of human umbilical cord mesenchymal stem cell transplantation on clinical treatment? Stem Cell Res Ther. 2020;11(1):1–13.
Fan C-G, Zhang Q-J, Zhou J. Therapeutic potentials of mesenchymal stem cells derived from human umbilical cord. Stem Cell Rev Rep. 2011;7(1):195–207.
Gerwin N, Bendele A, Glasson S, Carlson CJO. The OARSI histopathology initiative-recommendations for histological assessments of osteoarthritis in the rat. Osteoarthr Cartil. 2010;18:S24–34.
Conaghan PG, Hunter DJ, Cohen SB, Kraus VB, Berenbaum F, Lieberman JR, Jones DG, Spitzer AI, Jevsevar DS, Katz NP, et al. Effects of a single intra-articular injection of a microsphere formulation of triamcinolone acetonide on knee osteoarthritis pain: a double-blinded, randomized, placebo-controlled, multinational study. J Bone J Surg Am. 2018;100(8):666.
Moody HR, Heard BJ, Frank CB, Shrive NG, Oloyede AO. Investigating the potential value of individual parameters of histological grading systems in a sheep model of cartilage damage: the Modified Mankin method. J Anat. 2012;221(1):47–54.
De Picker L, Fransen E, Coppens V, Timmers M, de Boer P, Oberacher H, Fuchs D, Verkerk R, Sabbe B, Morrens MJ. Immune and neuroendocrine trait and state markers in psychotic illness: decreased kynurenines marking psychotic exacerbations. Front Immunol. 2020;10:2971.
Yea JH, Bae TS, Kim BJ, Cho YW, Jo CH. Regeneration of the rotator cuff tendon-to-bone interface using umbilical cord-derived mesenchymal stem cells and gradient extracellular matrix scaffolds from adipose tissue in a rat model. Acta Biomater. 2020;114:104–16.
Kusuma GD, Carthew J, Lim R, Frith JE. Effect of the microenvironment on mesenchymal stem cell paracrine signaling: opportunities to engineer the therapeutic effect. Stem Cells Dev. 2017;26(9):617–31.
Son TW, Yun SP, Yong MS, Seo BN, Ryu JM, Youn HY, Oh YM, Han HJ. Netrin-1 protects hypoxia-induced mitochondrial apoptosis through HSP27 expression via DCC- and integrin α6β4-dependent Akt, GSK-3β, and HSF-1 in mesenchymal stem cells. Cell Death Dis. 2013;4(3): e563.
Liang Y, Chen S, Yang Y, Lan C, Zhang G, Ji Z, Lin H. Vasoactive intestinal peptide alleviates osteoarthritis effectively via inhibiting NF-κB signaling pathway. J Biomed Sci. 2018;25(1):1–8.
Haraden CA, Huebner JL, Hsueh M-F, Li Y-J, Kraus VB. Synovial fluid biomarkers associated with osteoarthritis severity reflect macrophage and neutrophil related inflammation. Arthritis Res Ther. 2019;21(1):1–9.
Zayed M, Schumacher J, Misk N, Dhar MJ. Effects of pro-inflammatory cytokines on chondrogenesis of equine mesenchymal stromal cells derived from bone marrow or synovial fluid. Vet J. 2016;217:26–32.
Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol. 2021;11:584116.
Cascão R, Moura RA, Perpétuo I, Canhão H, Vieira-Sousa E, Mourão AF, Rodrigues AM, Polido-Pereira J, Queiroz MV, Rosário HS, et al. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res Ther. 2010;12(5):1–8.
Liu X, Li X, Tao Y, Li N, Ji M, Zhang X, Chen Y, He Z, Yu K, Yu Z. TCDD inhibited the osteogenic differentiation of human fetal palatal mesenchymal cells through AhR and BMP-2/TGF-β/Smad signaling. Toxicology. 2020;431: 152353.
Abney KK, Galipeau J. Aryl hydrocarbon receptor in mesenchymal stromal cells: new frontiers in AhR biology. FEBS J. 2021;288(13):3962–72.
Podechard N, Fardel O, Corolleur M, Bernard M, Lecureur V. Inhibition of human mesenchymal stem cell-derived adipogenesis by the environmental contaminant benzo(a)pyrene. Toxicol In Vitro. 2009;23(6):1139–44.
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203.
Acknowledgements
This study was supported by National Natural Science Foundation of China (Nos. 81573443, 82173824, 81973332, 82003763); Anhui Province Natural Science Fund (outstanding youth) (No. 170808J10); Natural Science Foundation of Anhui Provincial (No. 2108085MH320, 2108085QH383); Collaborative Innovation Project of Key Scientific Research Platform in Anhui Universities (No. GXXT-2020-065); The Open Fund of Key Laboratory of Anti-inflammatory and Immune Medicine, Ministry of Education, P.R. China (Anhui Medical University) (No. KFJJ-2020-05); Improvement Program of Scientific Research Basement Construction (2021xkjT043); Scientific Research Fund of Anhui Medical University (No. 2020xkj026).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethics approval
All protocols were authorized by the Ethics Committee of Anhui Medical University (No. LLSC-20200992).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, X., Zhao, Y., Li, S. et al. Activation of the kynurenine–aryl hydrocarbon receptor axis impairs the chondrogenic and chondroprotective effects of human umbilical cord-derived mesenchymal stromal cells in osteoarthritis rats. Human Cell 36, 163–177 (2023). https://doi.org/10.1007/s13577-022-00811-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-022-00811-4