
Abstract
The SLC7A11/GPX4 axis plays an important role in ferroptosis during cardiac ischemia/reperfusion injury (IRI). The present study was designed to evaluate the impact of dexmedetomidine (DEX) post-conditioning on cardiac IRI and to explore whether the effect was achieved by SLC7A11/GPX4 signaling pathway regulation. Rat myocardial IRI was established by occluding the left anterior descending artery for 30 min followed by 2-h reperfusion. The infarct area was detected by diphenyltetrazolium chloride (TTC) staining; the cardiac function was evaluated by echocardiography. The levels of lipid peroxide biomarkers were measured to estimate the injury caused by lipid peroxide. HE staining and Sirius staining were utilized to assess myocardial damage and fibrosis. The mitochondrial morphology was observed by electron micrography. Western blot and quantitative real-time polymerase chain reaction were employed to measure the relative molecular characteristics. Our results showed that DEX administration at the beginning of reperfusion attenuated IRI-induced myocardial injury, alleviated mitochondrial dysfunction, decreased the level of reactive oxygen species (ROS), alleviated mitochondrial dysfunction, inhibited the activation of SLC7A11/GPX4, and modulated the expression of ferroptosis-related proteins, including SLC7A11, glutathione peroxidase 4 (GPX4), ferritin heavy chain (FTH), and cyclooxygenase-2 (COX-2). Conversely, the ferroptosis activator erastin partly suppressed the DEX-mediated cardio protection. Altogether, these results reveal that DEX inhibits ferroptosis by enhancing the expression of SLC7A11 and GPX4, thereby preventing cardiac I/R injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although myocardial reperfusion is the basis for saving myocardial viability, the restoration of the coronary blood flow presumably results in the death of myocardial cells. A previous study evidenced that volatile anesthetics decreased the myocardial oxygen demand and maintained its oxygen balance; thus, they possibly may play a crucial part in myocardial injury reduction [1]. However, this presumption is still only at the theoretical stage, and a huge gap divides experimental results from clinical applications.
Ferroptosis is a new form of regulated cell death, characterized by iron-dependent accumulation of lipid peroxides [2, 3]. It can be triggered by Xc-system inhibition and inactivation of glutathione peroxidase 4 (GPX4). Xc-system receives cystine from the extracellular environment and by reduction converts it into cysteine in the cytoplasm, which is then used for glutathione (GSH) synthesis [4, 5]. AsGPX4 is a GSH-dependent enzyme that can degrade small molecular peroxides, prevent lipid peroxidation, and decrease the sensitivity to ferroptosis [6]. In addition, SLC7A11 is the catalytic subunit of the Xc-system, whose upregulation can increase intracellular cystine levels, leading to GSH biosynthesis and thus enhancing the GPX4 activity but suppressing ferroptosis [7]. A recent study showed that ferroptosis is involved in various pathological processes, including acute kidney failure, liver injury, and heart disease, as well as myocardial IRI [8]. Baba et al. suggested that the mechanistic target of rapamycin (mTOR) prevented myocardial IRI through decreasing the accumulation of ferric ions and the production of lipid peroxides [9]. Fang et al. also revealed that ferroptosis mediated cardiomyopathy induced by chemotherapy and IRI [10].
As a selective α2-adrenergic receptor, DEX has been recommended for patients with sepsis more than 2 decades ago [11]. Clinical data have revealed that it inhibits the activity of sympathetic nerves and has analgesic and sedative effects [12, 13]; meanwhile, it also has protective activity on the brain and heart [14]. And a study found that DEX inhibited the TLR4/NF-κB pathway by activating α2A-adrenergic receptors, thus alleviating liver I/R injury [15]. Additionally, the pretreatment of DEX activated the PI3K/Akt signaling pathway in a α2 adrenoceptor-dependent manner, resulting in inhibition of the I/R-induced cell apoptosis and protection from cardiac injury [16]. Furthermore, our present results suggest that the attenuation of iron concentration as well as the inhibition of ferroptosis by enhancing GPX4 may be major mechanisms by which DEX alleviates myocardial injury [17]. In the present study, we hypothesized that DEX post-conditioning alleviated cardiac IRI in rats through the inhibition of ferroptosis. In addition, the involvement and underlying molecular mechanism of action of the SLC7A11/GPX4 axis were also explored.
Materials and methods
Animals
Healthy male Sprague–Dawley (SD) rats were provided from medical laboratory animal center of Jiangnan University and used at 230–250 g. All the animal tests were conducted in adherence with the guidelines for the Principles of Laboratory Animal Care and Use of Laboratory Animals published by NIH (NIH Publication, 8th Edition, 2011) and approved by the Ethics Committee for the Use of Experimental Animals in Jiangnan University (the approval number: JN.No20210315S1080910 [42]). The date for animals to be allowed was March 10, 2021.
Langendorff I/R model and hemodynamic measurements.
The rats were anesthetized with sodium pentobarbital (40 mg/kg) to the effect that the plantar and eyelid reflex would not appear in the experimental protocol, and heparin (1000 IU/kg) was injected intraperitoneally for anticoagulation [18, 19]. Then, the hearts were quickly excised and mounted on a Langendorff apparatus via the aorta for retrograde perfusion with Krebs–Henseleit (K–H) solution at constant pressure (10 kPa). K–H solution configuration (mmol/L): NaCl 118.0, KCl 4.8, KH2PSO4 1.2, NaHCO3 25.0, MgSO4 1.2, CaCl2 2.5, glucose 11.0, and pH 7.35–7.45. After pre-access 95% O2–5% CO2 for 30 min, K–H solution was maintained at 37 ℃ by an external circulation (Alcott Biotechnology Co, Ltd, Shanghai, China). The pump was adjusted to maintain coronary transudes to 12 mL/min.
A small latex balloon connected to a cuff pressure transducer (SIA Industrial & Trade, Beijing, China) was inserted into the left ventricle through the mitral valve to monitor the heart function. The cuff volume was adjusted to achieve a stable left ventricular end-diastolic pressure (LVEDP) of 5–10 mmHg during initial equilibration. Cardiac functions, including left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), ± dp/dtmax and heart rate (HR) were recorded at 30 min of equilibration (T0), 30 min (T1), 60 min (T2), 90 min (T3), and 120 min (T4) after reperfusion by Med Lab 6.0 software. Rats with refractory ventricular fibrillation, frequent arrhythmia, LVSP < 75 mmHg or HR < 180 beats/min were excluded.
Experimental protocols
One hundred and eight isolated perfused rat hearts were randomly divided into four groups: Control (CON) group, ischemia/reperfusion (I/R) group, dexmedetomidine post-conditioning (DePC) group, and dexmedetomidine + erastin post-conditioning (DePC + erastin) group. Excepting the Control group, rat hearts were subjected to 30 min ischemia followed by 120 min reperfusion. DePC group received 3 nM dexmedetomidine for the first 15 min of reperfusion. In DePC + erastin group, 10 μM ferroptosis inducer erastin was co-administered with 3 nM dexmedetomidine for 15 min at the onset of reperfusion. The experimental design is shown in Fig. 1.

Schematic diagram of the experiment. The red area represents ischemia time; the black area represents reperfusion time; the yellow area represents 2.5% of DEX administered for 15 min; the green area represents 5 mg/kg Erastin administered for 30 min (i.p.)
Determination of infarct size
Once the reperfusion reached an end, we ligated the left coronary immediately followed by the administration of Evans blue (10 mM) into the left ventricular cavity in vivo. After the perfusion was throughout the non-ischemic heart, Evans blue-stained parts were removed and the rest left ventricle was sliced into 1 mm-thickness pieces. The slices were dyed by 1% diphenyltetrazolium chloride (TTC) solution at 37 ℃ for 15 min without light and the active myocardial was represented as brick red. Then they were fixed for 24 h in a 4% paraformaldehyde solution. Finally, measurement by image software (IMAGE J, NIH, USA) was performed in the infarct area. The infarct size was defined as the ratio of the infarct area to the risk one.
Measurement of myocardial creatine kinase-MB (CK-MB), lactate dehydrogenase (LDH), and cardiac troponin I (cTnI)
After collecting the blood samples, they were separated by centrifugation at 3000 rpm. The measurement of myocardial LDH, myocardial creatine kinase-MB (CK-MB) and cardiac troponin (cTnl) were performed spectrophotometrically (Lightway PQY-01, JP) using commercial kits in a blinded manner according to the manufacturer's instruction (Jiancheng Bioengineering Institute, Nanjing, China).
Quantitative real-time PCR
Total RNA was extracted from left ventricular tissues using the Trizol reagent (TakaRa, Kyoto, Japan) and the concentration of RNA was measured with the use of Nano Drop 2000 (Thermo Science). According to the instruction, 0.5 μg RNA was utilized for each sample to reverse transcribe into cDNA and then was stored at − 20 ℃. The PCR condition was as follows: the polymerase chain reaction initiated at 95 ℃ for 15 s, followed by 40 cycles of 20 s of denaturation at 95 °C, an annealing at 55 °C for 30 s and an extension at 70 °C for 30 s. All measurements were quantified by the expression of DAPDH, and the multiplication was calculated by 2−ΔΔCT method.
Morphological observation
Hematoxylin and eosin (HE) staining and Sirius red were performed for the assessment of morphological changes by microscopy. The left ventricular myocardial tissues were fixed in 4% paraformaldehyde at room temperature and embedded in paraffin. Then they were mounted into sections and deparaffinized with the use of xylene. After dewaxing, the slides were stained with hematoxylin–eosin and Sirius red for observation of pathological changes.
Immunohistochemistry for 4-HNE
The paraffin sections were fixed with formaldehyde and incubated in 3% hydrogen peroxide for 30 min to block the activity of endogenous peroxidase. Then they were cultivated with the primary mouse antibody against 4-hydroxynonenal (4-HNE, ab48506, Abcam, Cambridge, MA, USA) overnight. For antigen recovery, the sections were immunoassayed using the VECTASTAIN ABC kit according to the manufacturer's instructions. Diaminobenzidine is used for staining and visualization, and hematoxylin is used to stain sections.
Measurement of Fe2+
The left ventricular tissues were homogenized with phosphate buffered saline (PBS) and centrifugation. After that, the supernatant was used for iron concentration detection by Iron Assay Kit (Abcam, Shanghai, China) according to the manufacturer’s instruction.
Oxidative stress measurements
The left ventricular tissue homogenate was collected for the measurement of oxidative stress. In terms of ROS detection, AutoFluo quencher was added to the frozen sections of heart tissue to prevent autofluorescence. The frozen sections were then incubated with ROS fluorescence probe away from light at 37 °C for 30 min. After adding diamino phenylindole (DAPI) to stain cell nucleus, we used Olympus DX51 fluorescence microscope (Tokyo, Japan) to evaluate the level of myocardial ROS. The relative concentration of GSH and GSSH was detected with a Glutathione Assay Kit (CS0260; Sigma, USA). The relative concentration of malondialdehyde (MDA) in cell lysates was assessed with a Lipid Peroxidation Assay Kit (ab118970; Abcam). All kits were used according to the manufacturers’ instructions.
Electron microscopy
Fresh tissue samples were rapidly collected with caution from the left ventricle of rats. After placing in a tube containing stationary liquid for 4 h, the heart tissue underwent infiltration, dehydration and embedding overnight. Then, the sample was captured by a transmission electron microscope (Hitachi, Japan) and ultrastructural damage of mitochondria was evaluated according to the Flaming score.
Western blotting
Western blotting was performed as previously described. Briefly, after collection of the supernatants of the left ventricular tissue, protein samples (20–25 mg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were probed with primary antibodies, including GPX4 (ab125066, 1:1000, Abcam, Cambridge, MA, USA), SLC7A11 (98051, 1:500, Cell Signaling Technology, Danvers, MA, USA), Cox-2 (ab162331, 1:1000, Abcam, Cambridge, MA, USA), FTH (4393, 1:500, Cell Signaling Technology, Danvers, MA, USA), Diluted secondary antibodies were used to detect the corresponding primary antibodies. Further analysis was carried out using Image Pro Plus v6.0 (Media Cybernetics, Carlsbad, CA, USA) to quantify the protein bands.
Statistical analysis
The datum was applied as mean ± standard deviation. In addition, the comparison between two groups was analyzed by Student’s test. P < 0.05 indicated statistical significance. The statistical analysis was performed using SPSS 21.0 and Image Pro Plus v6.0.
Results
DEX attenuated I/R-induced myocardial injury and cardiac dysfunction in rats but was abolished by ferroptosis activation
We administered 3 nM DEX and 10 μM erastin in an isolated myocardium without ischemia treatment to establish whether exerted toxic effects on the normal myocardium. As can be observed in Figure S2, no differences were present in the myocardial infarction size and LDH content among the three groups. Further, our HE staining results showed that the aforementioned drugs had no significant effect on the normal myocardium as they did not lead to histopathological changes. The results of our concentration gradient experiments revealed that the 10 μM erastin treatment group significantly abolished the cardioprotection of DEX as compared to the erastin-untreated group. As visible in Fig. 2a, the myocardial infarct size was significantly higher in the DePC + 10 μM and 20 μM erastin-treated groups than in the DePC group. The WB results showed that the treatment with 10 and 20 μM erastin significantly inhibited the expression of GPX4. Thus, we chose 10 μM erastin for ferroptosis induction in the subsequent experiments.

DEX post-conditioning attenuated I/R-induced myocardial injury and cardiac dysfunction in rats but was abolished by ferroptosis activation. a Representative image of TTC staining after reperfusion. The red area represents normal myocardial tissue, and the lighter area is the infarcted and ischemic area. b Myocardial infarction area (percentage of area at risk), n = 6/group. c The levels of ANP, BNP, Myh7 were detected n = 6/group. d The levels of CK-MB, LDH, and cTnI were detected, n = 6/group. *P < 0.05; the value is expressed as mean ± SD
To further characterize the role of DEX post-conditioning in myocardial I/R injury, we applied it 15 min post-ischemia. As illustrated in Fig. 2a, b, I/R injury increased the myocardial infarction size (P < 0.05), whereas DEX decreased the I/R-induced myocardial infarction (P < 0.05). Hence, to explore whether ferroptosis was involved in cardiac protection, isolated hearts were treated with erastin, a highly effective ferroptosis inducer, in addition to the treatment with DEX. The obtained results revealed that the erastin treatment led to a robust increase in the myocardial infarction area (P < 0.05).
Another powerful approach of evaluating myocardial injury is detecting the mRNA levels of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and slow/beta cardiac myosin heavy-chain (Myh7), three classic biomarkers of cardiac hypertrophy. Similarly to the aforementioned results, we also discovered that the treatment with DEX considerably prevented the upregulation of the mRNA levels induced by I/R injury. Additionally, the mRNA levels of ANP, BNP, and Myh7 were more mildly enhanced by the treatment with both DEX and erastin than those in the single DEX treatment group (Fig. 2c, P < 0.05). Meanwhile, we detected marker enzymes which are closely associated with infarction size and myocardial injury to further confirm the DEX-induced protection. As can be seen in Fig. 2d, I/R injury increased the levels of LDH, CK-MB, and cTnl, whereas their levels were downregulated by the treatment with DEX. Nonetheless, the LDH, CK-MB, and cTnl levels were significantly more elevated in the DePC + erastin group than in the DEX group (P < 0.05).
Further, we monitored and recorded the hemodynamics indexes in all groups at T0, T1, T2, T3, and T4 to assess cardiac function. At T1, T2, T3, and T4, HR and LVSP had lower values than those at T0, whereas that of LVEDP was higher in all groups, except for the Con group (P < 0.05, Table 1). In addition, when compared with the Con group, it represented a descent in HR and LVSP along with an increased LVEDP at T1, T2, T3, and T4 in the other groups (P < 0.05, Table 1). As expected, DEX post-conditioning remarkably restrained the cardiac dysfunction resulting from myocardial I/R injury (P < 0.05, Table 1). However, HR and LVSP were lower while LVEDP was higher in the DePC + erastin group (P < 0.05, Table 1) than the respective values in the DePC group. Taken together, these results suggested that ferroptosis can abolish the cardioprotective effect of DEX on I/R-induced myocardial injury and cardiac dysfunction.
DEX alleviated myocardial structural lesions, preventing against I/R-induced ferroptosis
Cardiac I/R damage often leads to structural damage and formation of pathological scar. As shown in Fig. 3a, b, HE and Sirius red staining in the control group showed that the myocardial tissue structure was intact, the cardiomyocytes were arranged normally, and no obvious fibrosis were observed. Conversely, the myocardial tissue in the I/R group was disorderly arranged, and the infarct area was loose, with edema similar to that in the sham operation group. In contrast, disordered alignment of myofilaments and sarcomeres were observed in the I/R group. We observed scattered hypertrophic cardiomyocytes, fibroblast proliferation, large area of collagen fiber deposition (mainly type I and type II collagen fibers), and fibrous scar formation. The administration of DEX led to a noticeably clearer myocardial tissue structure than that in the I/R group; the myocardial damage was reduced, and the areas of fibrosis were smaller in the DEX treatment group. Nevertheless, after erastin treatment, the myocardial structural damage and fibrosis area increased significantly, indicating that with the activation of ferroptosis, the myocardial protective effect of DEX was offset.

DEX post-conditioning alleviated myocardial structural lesions against I/R-induced ferroptosis. a Representative image of HE-stained heart slice, magnified ×200, scale bar = 100 μm. b Representative image of a heart slice stained with Sirius red, magnified ×200, scale bar = 100 μm
DEX counteracted the production of cell-damaging free radicals and was involved in oxidative stress mediation by erastin
To determine whether DEX has a protective effect against cardiac I/R injury through an antioxidant mechanism of action, we assessed the expression of the lipid degradation product 4-hydroxynonenal (4-HNE), a secondary product of lipid peroxidation. As can be seen in Fig. 4a, the relative 4-HNE level in the Con group was significantly lower than those in the other groups. Our results showed a more significant decrease in the DePC group than in the I/R group whereas these levels in were further increased in the DePC + erastin group. Interestingly, elevated ROS is not only a significant cause of ferroptosis, but also its outcome. Here, we found that DEX alleviated the increase in the cardiac ROS levels induced by IR injury (Fig. 4b , P < 0.05). The administration of erastin significantly activated the ferroptosis in the DePC + erastin group, manifested as a higher ROS production than that in the DEX group (Fig. 4c, P < 0.05).

DEX improves cardiac IRI by inhibiting oxidative stress. a Representative immunohistochemical staining, magnification 200, scale bar = 100 μm. b Quantitative analysis relative to 4-HNE level, = 6/group, = 4/group. c Stain cell nucleus ROS was stained by DAPI. d Quantitative analysis of relative ROS levels, = 6/group, = 4/group. e–h Biomarkers of oxidative stress were assessed, myocardial GSH, GSSG, GSH/GSSG and MDA levels, = 6/group. *P < 0.05; the value is expressed as mean ± SD
In addition, we evaluated biomarkers of oxidative stress, including GSH, GSSH, and MDA (Fig. 4e–h, P < 0.05). In all experimental groups, the levels of GSSH and MDA were significantly higher but the levels of GSH and GSH/GSSH were significantly than those in the control group. Additionally, the level of GSH in the DePC group was significantly higher than that in the I/R group, but the GSSH and MDA levels in the DePC group were lower. It is noteworthy that the treatment with erastin significantly increased the levels of the biomarkers of oxidative stress; the erastin application significantly reduced the levels of GSH, whereas it remarkably increased those of GSSH and MDA.
DEX improved the mitochondrial structure and function, but this effect was inhibited by erastin treatment
As previous studies have evidenced, abnormal mitochondrial structure and function and oxidative damage may induce ROS overproduction [14]. Therefore, to further validate our hypothesis, the cardioprotective effect of DEX was evaluated by an assessment of the structure and function of the mitochondria and the TOM20 myocardial ultrastructure and combustion score. As can be seen in Fig. 5, the cardiomyocytes of the control group were arranged neatly; the sarcomere and intercalated disk are arranged neatly, the mitochondria had normal appearance and structure, without swelling, and the cristae density was intact. Compared with the control group, the ultrastructural damage in the I/R group was more pronounced. The myocardial tissue had defects of sarcomere and edematous rupture, and the arrangement of myocardium was disordered. Obvious vacuolization and cristae rupture appeared in the mitochondria. The myocardial ultrastructure damage in the DEX group was significantly attenuated compared with that in the I/R group; the sarcomeres had an almost parallel arrangement, and the mitochondrial structure returned to normal. The treatment with DEX combined with erastin led to more significant myocardial ultrastructure damage in the DEX + erastin group than in the DEX group; the sarcomere arrangement was disordered, and the mitochondrial structure was obviously abnormal (Fig. 5a–c, P < 0.05). In addition, the burn scores of all groups showed similar trends (Fig. 5d, P < 0.05). The scores of all experimental groups increased more sharply than that of the control group. The burn score of the I/R group was the highest. Compared with the I/R group, the burn score of the DEX group was considerably improved. However, the treatment with erastin increased again the burn score of the DEX + erastin group.

DEX post-treatment has the effect of improving the structure and function of cardiomyocyte mitochondria, but is inhibited viaerastin treatment. a Mitochondrial structure and TOM20 myocardial ultrastructure were observed by Olympus DX51 fluorescence microscope. b Quantitative analysis of myocardial Tom20 fluorescence intensity, n = 6/group. c Representative transmission electron microscope under magnification, 20,000, scale bar = 100 μm. It is worth noting that mitochondria are marked as (*), n = 3/group. d Quantitative analysis of Flameng score, n = 6/group, *P < 0.05; the value is expressed as mean ± SD
DEX regulated the expression of ferroptosis-related proteins in myocardial I/R isolated heart model
Ferroptosis is uniquely regulated cell death, characterized by ROS accumulation induced by lipid peroxidation and excessive iron deposition. We performed analyses to detect the relative content of iron in the serum and cardiomyocytes, which is essential for the realization of the ferroptosis mechanism. As can be seen in Fig. 6a, b, it was very low in the Con group, whereas its quantity in the I/R group was remarkably high. The treatment with DEX significantly decreased the amount of iron compared with that of the I/R group. The administration of erastin significantly elevated the relative iron content, with a similar trend in both the serum and the cells.

DEX reduces the iron content in cardiomyocytes by enhancing the signal transduction of SLC7A11/GPX4, thereby inhibiting ferroptosis. a Quantitative analysis of Iron relative content in serum n = 6/group. b Quantitative analysis of iron relative content of cardiomyocytes n = 6/group. c Representative immunoblotting of cytoplasmic SLC7A11 was performed. The quantitative analysis of SLC7A11 relative levels n = 6/group. d A representative western blot of cytoplasmic GPX4 was performed. Quantitative analysis of GPX4 relative levels n = 6/group. e A representative western blot of cytoplasmic FTH. Quantitative analysis of FTH relative levels n = 6/group. f A representative immunization of cytoplasmic COX-2 Blot. Quantitative analysis of COX-2 relative levels n = 6/group, *P < 0.05; the value is expressed as mean ± SD
Previous study revealed that the upregulation of SLC7A11 increased the expression of GPX4, thus suppressing the process of ferroptosis [17]. Then, we determined the levels of SLC7A11 and GPX4 was by Western blot to establish whether the protective effect of DEX on I/R-induced damage was achieved through SLC7A11/GPX4. Our results showed that the expression levels of SLC7A11 and GPX4 were considerably downregulated in the I/R group. In addition, they were significantly higher in the DePC group than in the I/R group and further downregulated by the combined treatment with DEX and erastin. Taken together, the upregulation of SLC7A11 was indirectly followed by upregulation of GPX4, which inhibited the DEX-induced effect by ferroptosis suppression (Fig. 6c–d, P < 0.05). Additionally, we evaluated the levels of FTH and COX-2, two proteins also associated with ferroptosis. Through storage of excessive iron in cells, FTH plays a crucial part in iron metabolism. The results of FTH analyses showed the same trend as that of the iron content. COX-2 was considerably higher in the I/R group than in the Con group. As expected, it was appreciably decreased by the DEX treatment, whereas its level in the DePC + erastin group was higher than that of the DEX group (Fig. 6e–f, P < 0.05). Overall, SLC7A11/GPX4 played an important role in DEX-induced protection through inhibiting ferroptosis.
Discussion
In this study, we used the Langendorff perfusion system to treat rats and obtain live cardiomyocytes. Our findings confirmed the myocardial protection and iron death inhibition effect of DEX. This is consistent with previous research results, such as those obtained by Wang, who found that DEX attenuated the myocardial iron myocardial iron overload and death caused by sepsis [17]. In earlier research, they found that DEX had a protective effect on myocardial ischemia and reperfusion [20]. Furthermore, the latest study indicated that DEX can prevent myocardial IRI by improving oxidative stress and apoptosis [21]. Most notably, all previous studies have confirmed that 0–100 nm DEX exerts a concentration-dependent protective effect on myocardial IRI. Consistent with previous studies, we found that 3 nm DEX had a protective effect on the isolated Langendorff myocardium. The latest research findings confirmed that 0.3–3 nm DEX post-conditioning improved the Langendorff-perfused heart in a concentration-dependent manner [47]. A further increase in the concentration did not enhance the protective effect of DEX post-conditioning. Therefore, we selected to examine the effect of 3 nm DEX post-conditioning and found that DEX reduced the myocardial infarct size, alleviated the mitochondrial dysfunction, decreased the ROS level, and downregulated the expression of SLC7A11/GPX4. An earlier investigation found that the rise in the level of the ferroptosis inducer erastin caused myocardial ischemia or aggravation of myocardial IRI [22]. We used erastin in our experiments and established that it partially negated the protective effect of DEX on the myocardium.
DEX is a highly specific α-2 receptor agonist with outstanding anxiolytic and sedative effects, without causing respiratory depression and with very few adverse effects. Therefore, it has been commonly used in clinical anesthesia [23]. In their research, Xue et al. speculated that DEX pre-conditioning may stimulate the α2-adrenergic receptors by inducing the TLR4/MyD88/NF-κB signaling pathway to prevent the lungs from IRI [24]. Another previous study revealed that DEX pre-conditioning improved the cardiac blood flow, reduced the infarct size, and protected the myocardial cells through activating multiple signaling pathways, such as Erk 1/2, Akt, and eNOS [25]. Other research findings also evidenced that DEX reduces the heart damage caused by CLP by increasing superoxide dismutase (SOD) and GSH levels [20]. Recently, Peng et al. showed that DEX post-condition attenuated both cardiac IRI in vivo and hypoxia/reoxygenation injury in vitro by targeting the HIF-1α signal pathway [19]. Notably, in another study, DEX not only promoted the functional recovery of the damaged myocardium, but also prevented the occurrence of ventricular fibrillation caused by IRI [26]. The aforementioned protective effect is possibly exerted via the stimulation of presynaptic α2-adrenergic receptors by the falling level of norepinephrine in myocardial cells [17, 27]. Recent evidence showed that DEX can improve myocardial ferroptosis caused by sepsis [17]. DEX was found to maintain iron homeostasis through the JNK/Sp1 and Stat4/Sp1 signaling pathways, thereby protecting SK-N-SH nerve cells from oxidative damage [28]. Similarly, our research revealed that DEX can attenuate iron death in myocardial IRI.
Ferroptosis is a type of programmed death caused by oxidative damage. Its mechanism is significantly morphologically, genetically, and biologically different from those of other forms of regulatory cell death, such as apoptosis, necrosis, and autophagy. The increased production of ROS and inactivation of GPX4 in the cell, as well as increased accumulation of lipid peroxides, leads to iron-dependent programmed cell death [3, 29]. Evidence has shown that ferroptosis plays a major role in the pathogenesis of cardiomyopathy. Ferroptosis was established to participate in by doxorubicin- and IRI-induced heart injury through the nuclear factor-E2-related factor (NRF2)/heme oxygenase-1 (Hmox1) axis [10]. DOX induces an increase in the mitochondrial content of iron in the cardiomyocytes, which damages the mitochondrial membrane. The accumulation of iron in the serum and heart tissue is caused by the upregulation of heme to free iron through Hmox1 and is independent of the classic hepcidin–ferroportin iron regulatory axis. In a mouse model of myocardial ischemia/reperfusion injury, the inhibition of ferroptosis and iron chelation has cardioprotective effects. This is consistent with the findings that the mitochondrial-targeting antioxidant MitoQ drastically improved I/R-related cardiac dysfunction, cell death, and mitochondrial damage [30]. Pharmacologically inhibiting ferroptosis or iron chelation therapy could remarkably prevent cardiomyopathy. Ferroptosis also participates in diabetic myocardial IRI damage by regulating the endoplasmic reticulum stress. The suppression of ferroptosis reduced the endoplasmic reticulum stress in an earlier study [23].
ROS can peroxidize lipids by reacting with the polyunsaturated fatty acids in the lipid membranes, leading to ferroptosis via the Fenton reaction [31]. Previous research has shown that Fer-1 pretreatment can inhibit ferroptosis by reducing the lipid ROS levels [32]. Ferroptosis occurrence is associated with the production of lipid ROS in all cell metabolism processes. There is evidence that the lipid and amino acid metabolism (especially the metabolism of cysteine and glutamine) is involved in the appearance of ferroptosis [3, 33]. This cell mechanism is related to significant morphological changes in the mitochondria, including mitochondrial fragmentation and cristae enlargement [34]. It was found that mitochondria have a key role in cysteine-deprivation-induced ferroptosis but not in that induced by GPX4 inhibition [35]. Other research has shown that DEX reduced the inflammation, oxidative stress, and apoptosis of cardiomyocytes by activating the AMPK-GSK3β signaling pathway [36]. Moreover, DEX pretreatment protected cardiomyocytes from IRI by inhibiting the NFκB signaling pathway and increasing ROS production [37]. ROS are produced in ischemic myocardium, especially after reperfusion, but the main source of ROS in the ischemic/reperfused myocardium is mitochondria [38]. Similarly, DEX can protect cardiomyocytes from oxidative stress damage and apoptosis by regulating the apoptosis signal pathway mediated by mitochondria and endoplasmic reticulum stress [39]. In this study, we found that DEX improved the mitochondrial condition and attenuated the negative effects of ROS, ultimately exerting myocardial protection.
SLC7A11, a key component of the cystine-glutamate antiporter, participates in GPX4-associated activities and is also considered the most crucial marker of ferroptosis in clinical practice [39,40,41]. SLC7A11, the light chain of the Xc-system, is used to transport cystine into the cell and glutamate out of the cell in a 1:1 exchange ratio [42]. Cystine is necessary for the synthesis of glutathione and is one of the essential cofactors of endogenous antioxidants [31]. Ferroptosis agonists can significantly reduce the expression of GPX4 in cardiomyocytes, leading to imbalance of iron metabolism and lipid peroxidation in cardiomyocytes [43]. The downregulation of SLC7A11 also results in a decrease in the intracellular cystine levels and a subsequent decrease in glutathione biosynthesis, which in turn inhibits GPX4 activity, increases ROS production, and destroys the integrity of the mitochondrial structure. Guan et al. evidenced that activating LC7A11/GPX4 axis exerted protection from I/R brain injury by ferroptosis inhibition [44]. Our study also confirmed that the SLC7A11/GPX4 axis can reduce myocardial IRI by suppressing ferroptosis. The conversion of Fe2+ to Fe3+ by the Fenton reaction contributes to lipid peroxidation and ROS production, which can have destructive impact on the cardiomyocytes [45, 46]. By FTH assessment, our study confirmed the occurrence of iron overload in hearts suffering from ferroptosis. However, DEX attenuated the adverse effects induced by ferroptosis.
Conclusion
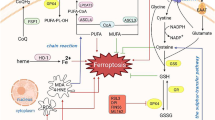
Our present results reveal that DEX provides cardioprotection against IRI. Our research findings showed that DEX inhibits ferroptosis by suppressing ROS production and maintaining the structural integrity of the mitochondria, thereby preventing myocardial IRI. As can be seen in Fig. 7, the mechanism of DEX protection on the myocardium may be exerted through the inhibition of the adverse effects of lipid ROS by activating the SLC7A11/GPX4 signaling pathway. As a result, mitochondrial damage is prevented, and the occurrence of ferroptosis in cardiomyocytes is reduced. The findings of this study support the hypothesis that treatments that act on the SLC7A11/GPX4 signaling pathway may be an effective strategy to reduce IRI in cardiomyocytes.

The schematic diagram of DEX up-regulates the SLCA11/GPX4 signaling pathway, inhibits Lipid ROS, reduces the occurrence of ferroptosis in cardiomyocytes, and thus has the protective effect of cardiac IRI. Solid green arrows depict promotion, positive regulation or activation. Transverse red “T” shape indicates inhibition, negative regulation or blockade. DEX dexmedetomidine, ROS reactive oxygen species, GSH glutathione, SLC7A11 solute carrier family 7, member 11, GPX4 glutathione peroxidase 4, COX-2 cyclooxygenase-2, FTH ferritin
Data availability statement
Research data are not shared.
References
Landoni G, Bignami E, Oliviero F, Zangrillo A. Halogenated anaesthetics and cardiac protection in cardiac and non-cardiac anaesthesia. Ann Card Anaesth. 2009;12:4–9.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Stockwell BR, FriedmannAngeli JP, Bayir H, Bush AI, Conrad M, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Liu MR, Zhu WT, Pei DS. System Xc-: a key regulatory target of ferroptosis in cancer. Investig New Drugs. 2021;39:1123–31.
Song X, Zhu S, Chen P, Hou W, Wen Q, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc-activity. Curr Biol CB. 2018;28:2388-2399.e5.
Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–70.
Gong Y, Wang N, Liu N, Dong H. Lipid peroxidation and gpx4 inhibition are common causes for myofibroblast differentiation and ferroptosis. DNA Cell Biol. 2019;38:725–33.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosisin cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314:H659–68.
Fang X, Wang H, Han D, Xie E, Yang X, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. 2019;116:2672–80.
Keating GM. Dexmedetomidine: a review of its use for sedation in the intensive care setting. Drugs. 2015;75:1119–30.
Taniguchi T, Kidani Y, Kanakura H, Takemoto Y, Yamamoto K. Effects of dexmedetomidine on mortality rate and inflammatory responses to endotoxin-induced shock in rats. Crit Care Med. 2004;32:1322–6.
Venn RM, Bradshaw CJ, Spencer R, Brealey D, Caudwell E, et al. Preliminary UK experience of dexmedetomidine, a novel agent for postoperative sedation in the intensive care unit. Anaesthesia. 1999;54:1136–42.
Kong W, Kang K, Gao Y, Liu H, Meng X, et al. Dexmedetomidine alleviates LPS-induced septic cardiomyopathy via the cholinergic anti-inflammatory pathway in mice. Am J Transl Res. 2017;9:5040–7.
Wang Y, Wu S, Yu X, Zhou S, Ge M, et al. Dexmedetomidine protects rat liver against ischemia-reperfusion injury partly by the α2A-adrenoceptor subtype and the mechanism is associated with the TLR4/NF-κB pathway. Int J Mol Sci. 2016;17:995.
Chang JH, Jin MM, Liu JT. Dexmedetomidine pretreatment protects the heart against apoptosis in ischemia/reperfusion injury in diabetic rats by activating PI3K/Akt signaling in vivo and in vitro. Biomed Pharmacother Biomed Pharmacother. 2020;127:110188.
Wang C, Yuan W, Hu A, Lin J, Xia Z, et al. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22:175–84.
Alakoski T, Ulvila J, Yrjölä R, Vainio L, Magga J, et al. Inhibition of cardiomyocyte Sprouty1 protects from cardiac ischemia-reperfusion injury. Basic Res Cardiol. 2019;114:7.
Peng K, Chen WR, Xia F, Liu H, Meng XW, et al. Dexmedetomidine post-treatment attenuates cardiac ischaemia/reperfusion injury by inhibiting apoptosis through HIF-1α signalling. J Cell Mol Med. 2020;24:850–61.
Zhong Y, Li YP, Yin YQ, Hu BL, Gao H. Dexmedetomidine inhibits pyroptosis by down-regulating miR-29b in myocardial ischemia reperfusion injury in rats. Int Immunopharmacol. 2020;86:106768.
Wu ZL, Davis J, Zhu Y. Dexmedetomidine protects against myocardial ischemia/reperfusion injury by ameliorating oxidative stress and cell apoptosis through the Trx1-dependent Akt pathway. BioMed Res Int. 2020. https://doi.org/10.1155/2020/8979270.
Li W, Li W, Leng Y, Xiong Y, Xia Z, et al. is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020;39:210–25.
Arcangeli A, D’Alò C, Gaspari R. Dexmedetomidine use in general anaesthesia. Curr Drug Targets. 2019;10:687–95.
Xue BB, Chen BH, Tang YN, Weng CW, Lin LN. Dexmedetomidine protects against lung injury induced by limb ischemia-reperfusion via the TLR4/MyD88/NF-κB pathway. Kaohsiung J Med Sci. 2019;35:672–8.
Ibacache M, Sanchez G, Pedrozo Z, Galvez F, Humeres C, et al. Dexmedetomidine preconditioning activates pro-survival kinases and attenuates regional ischemia/reperfusion injury in rat heart. Biochim Biophys Acta. 2012;1822:537–45.
Yoshitomi O, Cho S, Hara T, Shibata I, Maekawa T, et al. Direct protective effects of dexmedetomidine against myocardial ischemia-reperfusion injury in anesthetized pigs. Shock (Augusta, GA). 2012;38:92–7.
Mahboobi SK. Dexmedetomidine and renal protection after cardiac surgery. J Clin Anesth. 2020;40:121–2.
Qiu L, Ge L, Hu Q. dexmedetomidine protects SK-N-SH nerve cells from oxidative injury by maintaining iron homeostasis. Biol Pharm Bull. 2020;43:424–31.
Hao S, Yu J, He W, Huang Q, Zhao Y, et al. cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia (New York, NY). 2017;19:1022–32.
Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J Off Publ Feder Am Soc Exp Biol. 2005;19:1088–95.
Xie Y, Hou W, Song X, Yu Y, Huang J, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Li N, Wang W, Zhou H, Wu Q, Duan M, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radical Biol Med. 2020;160:303–18.
Gao M, Jiang X. To eat or not to eat-the metabolic flavor of ferroptosis. Curr Opin Cell Biol. 2018;51:58–64.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354-363.e3.
He Y, Yang Z, Li J, Li E. Dexmedetomidine reduces the inflammation and apoptosis of doxorubicin-induced myocardial cells. Exp Mol Pathol. 2020;113:104371.
He L, Wang Z, Zhou R, Xiong W, Yang Y, et al. Dexmedetomidine exerts cardioprotective effect through miR-146a-3p targeting IRAK1 and TRAF6 via inhibition of the NF-κB pathway. Biomed Pharmacother Biomed Pharmacother. 2021;133:110993.
Rajendran P, Nandakumar N, Rengarajan T, Palaniswami R, Gnanadhas E, et al. Antioxidants and human diseases. Clin Chim Acta Int J Clin Chem. 2014;436:332–47.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2020. https://doi.org/10.1080/15548627.2020.1810918.
Dong H, Qiang Z, Chai D, Peng J, Xia Y, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging. 2020;12:12943–59.
Fang X, Cai Z, Wang H, Han D, Cheng Q, et al. Loss of cardiac ferritin h facilitates cardiomyopathy via slc7a11-mediated ferroptosis. Circ Res. 2020;127:486–501.
Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x(c)(-): cystine supplier and beyond. Amino Acids. 2012;42:231–46.
Liu T, Jiang L, Tavana O, Gu W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Can Res. 2019;79:1913–24.
Guan X, Li Z, Zhu S, Cheng M, Ju Y, et al. Galangin attenuated cerebral ischemia-reperfusion injury by inhibition of ferroptosis through activating the SLC7A11/GPX4 axis in gerbils. Life Sci. 2021;264:118660.
Sumneang N, Siri-Angkul N, Kumfu S, Chattipakorn SC, Chattipakorn N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch Biochem Biophys. 2020;680:108241.
Sung HK, Song E, Jahng J, Pantopoulos K, Sweeney G. Iron induces insulin resistance in cardiomyocytes via regulation of oxidative stress. Sci Rep. 2019;9:4668.
Acknowledgements
This study was supported by grants from Jiangsu Province department of science and technology (BE2020634, BK20191138 to J.Z), Top Talent Support Program for young and middle-aged people of Wuxi Health Committee (BJ2020049 to J.Z), the Natural Science Foundation in Jiangxi Province grant (20192ACBL21037 to P.Y.), the National Natural Science Foundation of China (81760048 to J.Z. and 81760050 to P.Y.)
Author information
Authors and Affiliations
Contributions
PY, JZ, JRZ, YD, DDC, HJS, FLY, SYL, XZL, PPL, LHF, and SCY performed nearly all the experiments. PY, JZ, YD, and DDC performed hemodynamic study and analyzed the data. PY, JZ, and JRZ designed the whole study. PY, SYL, and XZL were the major contributors in writing the manuscript, and all the experiments were performed under their guidance. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, P., Zhang, J., Ding, Y. et al. Dexmedetomidine post-conditioning alleviates myocardial ischemia–reperfusion injury in rats by ferroptosis inhibition via SLC7A11/GPX4 axis activation. Human Cell 35, 836–848 (2022). https://doi.org/10.1007/s13577-022-00682-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-022-00682-9