
Abstract
MiR-663b has been demonstrated to be abnormally expressed in several cancer types and was involved in the progression of cancer. Although overexpression of miR-663b in colorectal cancer was observed, the role of miR-663b in colorectal cancer cells has not been identified yet. In this study, we analyzed expression of miR-663b in colorectal tumors and explored the molecular mechanism of miR-663b in colorectal cancer cells. MiR-663b was significantly overexpressed in colorectal tumors and cell lines. Downregulation of miR-663b inhibited cell proliferation and sphere forming ability in colorectal cancer cells. In addition, miR-663b downregulation inactivated Ras/Raf signaling activity and subsequently decreased YAP1 and CD44 expression in colorectal cancer cells. Using TargetScan software, TNK1, a negative regulator of Ras/Raf signaling, was predicted to be a target gene of miR-663b. Western blotting and RT-qPCR showed that TNK1 expression was negatively regulated by miR-663b. In addition, the direct binding of miR-663b to TNK1 mRNA was proved by dual luciferase reporter assay. Furthermore, downregulation of miR-663b inhibited colorectal cancer cell proliferation and stemness, which was reversed after siRNA-mediated silencing of TNK1. In conclusion, the current study revealed a pivotal role of miR-663b in the progression of colorectal cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Globally, colorectal cancer is the third most commonly diagnosed cancer [1]. Despite systematic therapy including surgery, chemotherapy and radiotherapy is developed for treatment of patients with colorectal cancer, the prognosis of patients with later stage colorectal cancer remains poor, and the 5-year overall survival of patients with stage IV colorectal cancer is slightly over 10% [2]. The unsatisfied prognosis is due to high metastatic capability and drug resistance during treatment [3]. It is known that the presence of colorectal cancer stem cells is responsible for tumor initiation, growth, metastasis and drug resistance [4]. Owing to substantial studies on the molecular mechanism of colorectal cancer, target therapy based on EGFR antibodies have been widely used as first-line therapy agents during treatment of metastatic colorectal cancer and greatly improved patient outcome [5].
The transmembrane receptor EGFR and the downstream Ras/Raf signaling is one of the most well-characterized cancer-related pathways [6]. Uncontrolled, sustainable activation of EGFR/Ras/Raf signaling leads to strong cell proliferation, survival and metastasis ability and play a pivotal role during cancer initiation, development and drug resistance [7,8,9]. In colorectal cancer, it is known that overexpression of EGFR elevates Ras/Raf signaling activity, promotes cancer cell proliferation and survival [10]. Initially discovered and cloned from differentiating murine embryonic stem cells, TNK1 is a non-receptor protein tyrosine kinase which negatively regulates Ras/Raf signaling [11, 12]. Knockout of TNK1 significantly activated Ras/Raf pathway and resulted in the development of spontaneous tumors including colorectal cancer in mice [13], indicating TNK1 was involved in the initiation of colorectal cancer. Methylation of TNK1 promoter may be responsible for TNK1 downregulation [13]. Until now, it is still not clear how TNK1 is regulated in colorectal cancer.
MicroRNAs (miRNAs) are small, non-coding, single-stranded RNAs that ubiquitously expressed in eukaryotic cells [14]. MiRNAs primarily exert their function via directly binding to 3′UTR of target gene mRNAs, leading to mRNA degradation or translation inhibition [15]. Individual miRNA can have hundreds of target genes and one specific gene can be regulated by numerous miRNAs depending on cell background. Expression of miRNAs is essential for almost all cell physiological processes [16]. Aberrant expression of miRNAs is implicated in abnormal cell proliferation, motility and diseases including cancer [17]. Microarray and next-generation sequencing based screen approaches have revealed numerous cancer-related genes and miRNAs. MiRNA profiles in a total of 1893 carcinoma/normal-paired samples and 290 adenoma tissue samples identified many differentially expressed miRNAs in colorectal cancer. Several miRNAs such as miR-150-5p and miR-650 have been experimentally validated as colorectal cancer-related miRNAs [18, 19]. According to the microarray data, miR-663b expression is twofold higher in colorectal cancer compared with normal colon tissues. MiR-663b was previously reported to promote ovarian cancer progression via targeting TUCS2 [20]. The serum levels of miR-663b were increased in patients with colorectal cancer compared with normal volunteers and its expression could act as an independent diagnostic biomarker for colorectal cancer [21]. However, the role of miR-663b in colorectal cancer remains unclear.
In the current study, we evaluated the role of miR-663b in colorectal cancer. We found overexpression of miR-663b in colorectal tumors and cell lines. Downregulation of miR-663b inhibited colorectal cancer cell proliferation and sphere forming. Our further study indicated that miR-663b directly targeted TNK1 to activate Ras/Raf signaling. Silencing of TNK1 rescued miR-663b downregulation induced cell proliferation inhibition and decreased sphere forming ability. The data demonstrated that miR-663b promoted colorectal cancer progression via TNK1/Ras/Raf signaling.
Materials and methods
Tissue collection
48 pairs of tumors and normal tissues were collected from patients with colorectal cancer during surgery in The First Hospital of Jilin University from June 2012 to September 2018. Patients included 31 males and 17 females, aged from 33 to 74 years. Accordingly to the clinical TNM staging system and histological grading [7, 22] were stage I, 22 were stage II, 11 were stage III and 8 were stage IV; 5 were grade 1, 26 were grade II and 17 were grade III. None of the patients received chemotherapy, radiotherapy or target therapy before surgery. The sample and survival information were obtained from The First Hospital of Jilin University. The protocol of the experiments was approved by the Human Research Ethic Committee of The First Hospital of Jilin University. All patients provided written informed consent. The tissues were stored at − 80 °C until required for RNA and protein extraction. All tissues were subjected to RNA extraction and RT-qPCR. A total of 12 pairs of tissues were subjected to protein extraction and western blotting.
Cell culture
Human colorectal cancer cell lines (HCT116 and HT29) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). These cell lines were maintained in DMEM (Gibco; Thermo Fisher Scientific, Waltham, MA) medium supplemented with 10% FBS. Cells were cultured in a 37 °C humidified incubator with 5% CO2.
Overexpression or downregulation of miR-663 in colorectal cancer cells
The miR-663b mimic, miR-663b inhibitor, miR-NC (negative control) mimic and miR-NC inhibitor were synthesized and purchased from RiboBio (Guangzhou, China). Lipofectamine 3000 was used to transfect miR-663 mimic or miR-663 inhibitor or the negative control into HCT116 and HT29 cells. On the first day, 2 × 106 cells were plated in six-well plates. On the next day, 20 nmol miR-663 mimic or miR-663 inhibitor or the negative control were mixed with 5 μL lipofectamine 3000 in 500 μL OptiMEM (Invitrogen; Thermo Fisher Scientific), incubated for 15 min and then added to the culture medium. After 48 h, cells were harvested and subjected to the following experiments.
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissues and cells using miRNeasy Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The RNA was quantified with the NanoDrop 2000 (Thermo Fisher Scientific), then reverse transcribed into first-stranded cDNA with M-MLV Reverse Transcriptase (Thermo Fisher Scientific). The cDNA was used to perform RT-qPCR with TB Green Fast qPCR Mix (TaKaRa, Tokyo, Japan) on a CFX-96 Realtime PCR System (Bio-Rad, Hercules, CA, USA). The PCR conditions included predenature at 95 °C for 30 s, denature at 95 °C for 5 s, prolong at 60 °C for 10 s (40 cycles). U6 and GAPDH were internal controls for miRNA and mRNA, respectively. Primer sequences: stem loop primer: 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACTTTTG-3′; miR-663b-forward: 5′-TCGGCAGGGGTGGCCCGGCCGT-3′; miR-663b-reverse: 5′-CTCAACTGGTGTCGTGGA-3′; U6-forward: 5′-ATTGGAACGATACAGAGAAGATT-3′; U6-reverse: 5′-GGAACGCTTCACGAATTTG-3′; TNK1-forward: 5′-TGAAAAGGCTACGTTCTGGGC-3′; TNK1-reverse: 5′-ACCCTCTGGGATCAGACACTT-3′; GAPDH-forward: 5′-CTGGGCTACACTGAGCACC-3′; GAPDH-reverse: 5′-AAGTGGTCGTTGAGGGCAATG-3′.
Protein extraction and western blotting
The protein lysates were prepared with RIPA lysis buffer (Thermo Fisher Scientific) added with a cocktail of proteinase inhibitors and phosphatase inhibitors (Cell Signaling Technology, Danvers, MA, USA). Antibodies of TNK1 (sc-390359, 1:2000) and ERK1/2 (sc-514302, 1:2000) were obtained from Santa Cruz Technology (Dallas, TX). P-MEK (#2338, 1:1000), MEK (#9122, 1:1000), P-ERK1/2 (#4370, 1:1000), YAP1 (#14074, 1:1000) and CD44 (#3570, 1:1000) antibodies were purchased from Cell Signaling Technology. GAPDH (AM4300, 1:5000) antibody was bought from Thermo Fisher Scientific. Secondary antibodies against mouse (#7076, 1:10,000) and rabbit (#7074, 1:10,000) were purchased from Cell Signaling Technology. The concentration of protein lysates was determined with the BCA Protein Assay Kit (Thermo Fisher Scientific). Lysates containing 20 μg proteins were loaded in the 8% SDS-PAGE gels and separated via electrophoresis. The proteins were transferred to the PVDF membranes and incubated with primary antibodies and secondary antibodies sequentially. Finally, the blots were developed with the ECL Western Blot Substrate (Pierce; Thermo Fisher Scientific). The relative expression of proteins was normalized to GAPDH and analyzed with Image J software.
Proliferation assay
The proliferation ability of cells was detected with the CCK-8 kit (DoJinDo, Toyoko, Japan). Briefly, 1 × 104 cells were plated in 96-well plates. At the time point of transfection, 1, 2 and 3 days after transfection, 10 μL CCK-8 solution was added into each well and maintained for 2 h. The absorbance at 450 nm was determined with a Microplate Reader (Bio-Rad) to reflect cell number.
Sphere forming assay
To investigate the sphere formation ability, HCT116 and HT29 cells were plated on 96-well ultralow-attachment plates (Corning, New York City, NY, USA) at a density of 500 cells per well. The cells were maintained in sphere-forming medium made with serum-free DMEM/F12 medium (Gibco) supplemented with 20 ng/mL basic fibroblast growth factor (bFGF, R&D Systems, Minneapolis, MN), 20 ng/mL epidermal growth factor (EGF, PeproTech, Chicago, IL, USA), ITS (insulin–transferrin–selenium; Sigma-Aldrich), and B-27 supplement (Gibco). 15 days after plating, tumor spheres were observed and images were captured with an inverted phase-contrast microscope (Olympus).
Flow cytometry assay
To detect the cell surface expression of CD44, harvested cells were blocked with Ca2+, Mg2+-free HBSS (Thermo Fisher Scientific) containing 1% goat serum (Thermo Fisher Scientific) for 30 min at 4 °C. Cells were then incubated with CD44 antibody (1:100) for 1 h at 4 °C. After that, cells were incubated with FITC-conjugated secondary antibody (#ab7057, 1:2000, Abcam) for 1 h at 4 °C. After washing with PBS, cells were subjected to flow cytometry analysis on a MACSQuant Analyzer 10 (Miltenyi, Bergisch Gladbach, Germany).
Bioinformatic analysis
The expression data of miR-663b in normal colon mucosa (n = 381), colon adenoma (n = 51) and colon carcinoma (n = 411) were retrieved from GSE115513 dataset and analyzed by online tool GEO2R using GEO (https://www.ncbi.nlm.nih.gov/geo/). The potential target genes of miR-663 were analyzed on the TargetScan software (http://www.targetscan.org/vert_72/). There were 3642 target genes predicted for miR-663. The top 100 was analyzed. After literature review, we noticed that TNK1, a key regulator of Ras signaling and implicated in colorectal cancer, was a potential target gene of miR-663.
Dual luciferase reporter assay
The 3′UTR of TNK1 containing putative binding site for miR-663b was amplified from HCT116 cDNA and cloned into pGL3-basic plasmid. The mutant TNK1 3′UTR was constructed with a QuikChange Site-Directed Mutagenesis Kit (Agilent Technology, Santa Clara, CA, USA). The pGL3-TNK1 3′UTR-WT or pGL3-TNK1 3′UTR-Mut was co-transfected with miR-663 mimic or miR-NC mimic into cells using Lipofectamine 3000. 48 h after transfection, the relative luciferase activity was determined with a Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA).
Statistical analysis
All data were analyzed with GraphPad Prism 6.0 software. The data were presented as mean ± SD. All experiments were repeated three times. Differences between two groups were compared with Student’s t test. Differences among three groups were compared with one-way ANOVA followed by Newman–Keuls analysis. Kaplan–Meier analysis of overall survival was performed on the 48 patients with colorectal cancer. Log-rank test was used for comparison between groups. The differences were considered significant if p values were less than 0.05.
Results
MiR-663b expression was decreased in colorectal cancer and associated with poor prognosis
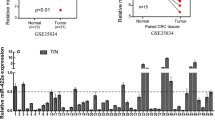
To explore the potential tumor-associated miRNAs in colorectal cancer, normal colon mucosa (n = 381), colon adenoma (n = 51) and colon carcinoma (n = 411), samples, previously analyzed for miRNA expression pattern, were bioinformatically re-analyzed for differentially expressed miRNAs. MiR-663b was identified as an upregulated miRNA in both colon adenoma and colon carcinoma compared with normal colon mucosa (Fig. 1a). For validation, we collected 48 pairs of colorectal tumors and matched normal tissues. RT-qPCR data consistently showed that miR-663b was significantly overexpressed in colorectal tumors (Fig. 1b). Additionally, according to our obtained survival information, patients with high expression of miR-663b exhibited a relatively poorer overall survival compared to patients with low expression of miR-663b (Fig. 1c).

The expression of miR-663b was elevated in colorectal tumors. a MiRNA expression profile of normal colon mucosa (n = 381), colon adenoma (n = 51) and colon carcinoma (n = 411) was downloaded from GSE115513. Bioinformatic analysis indicated that miR-663b was increased in colon adenoma and colon carcinoma compared with colon mucosa. b RT-qPCR was performed to detect miR-663b expression in 48 pairs of colorectal tumors and normal tissues. The data suggested that miR-663b was overexpressed in colorectal tumors. c Kaplan–Meier analysis of overall survival of colorectal patients with high and low expression of miR-663b. Patients with high expression of miR-663b showed shorter overall survival compared with those with low expression of miR-663b. ***p < 0.001. All experiments were repeated three times
MiR-663b promoted colorectal cancer cell proliferation and stem-like property
We next evaluated miR-663b expression in a panel of colorectal cancer cell lines (HCT116, HT29, SW620) and the immortal colon cells (FHC). RT-qPCR showed that miR-663b expression was increased in all colorectal cancer cell lines (HCT116, HT29, SW620) when compared with the immortal colon cells (FHC) (Fig. 2a). According to the expression pattern of miR-663b, we chose HCT116 and HT29 for further study. We downregulated miR-663b expression in HCT116 and HT29 cells by transfection of miR-663b inhibitor (Fig. 2b). Cell proliferation assay showed that downregulation of miR-663b inhibited cell proliferation in HCT116 cells (Fig. 2c). Consistently, the proliferation of HT29 cells was also strongly inhibited towards miR-663b downregulation (Fig. 2d). Cancer stemness was tightly associated with recurrence, drug resistance of colorectal cancer. We investigated the impact of miR-663b on the stem-like property of colorectal cancer cells. It was found that downregulation of miR-663b decreased the number of tumor spheres formed by HCT116 cells under anchorage-independent and serum-free conditions (Fig. 2e). Also, downregulation of miR-663b dramatically decreased the number of tumor spheres formed by HT29 cells (Fig. 2f). Together, these data suggested that miR-663b expression was essential for colorectal cell proliferation and stemness.

Downregulation of miR-663b inhibited cell proliferation and sphere forming in colorectal cancer cells. a RT-qPCR showed that miR-663b was overexpressed in HCT116, HT29 and SW620 cells when compared with FHC cells. b Transfection of miR-663b inhibitor decreased miR-663b expression in HCT116 and HT29 cells. c Downregulation of miR-663b decreased cell viability in HCT116 cells. d Downregulation of miR-663b decreased cell viability in HT29 cells. e Downregulation of miR-663b decreased number of tumor sphere of HCT116 cells. f Downregulation of miR-663b decreased the number of tumor spheres of HT29 cells. **p < 0.01; ***p < 0.001. All experiments were repeated three times
MiR-663b downregulation decreased the activity of Ras/Raf signaling and YAP1 expression
Ras/Raf is a well-characterized oncogenic signaling in various cancer types including colorectal cancer [23]. Western blotting results showed that the phosphorylated MEK and ERK1/2 were decreased in HCT116 and HT29 cells treated with miR-663b inhibitor (Fig. 3a). The ratio between phosphorylated MEK/total MEK and phosphorylated ERK1/2/total ERK1/2 was significantly decreased upon miR-663b downregulation (Fig. 3b), suggesting inactivation of Ras/Raf signaling. Ras/Raf signaling is involved in regulating of cancer stemness via elevation of YAP1 and CD44 [24, 25]. Downregulation of miR-663b decreased YAP1 and CD44 protein expression in HCT116 and HT29 cells (Fig. 3c, d). CD44 was a key mediator of cancer stemness expressed on the cell surface. Using flow cytometry, we observed that the surface expression of CD44 was decreased in both HCT116 and HT29 cells transfected with miR-663b inhibitor (Fig. 3e, f).

Downregulation of miR-663b inactivated Ras/Raf signaling in colorectal cancer cells. a Downregulation of miR-663b decreased p-MEK and p-ERK1/2 protein expression in HCT116 and HT29 cells. b Quantification of ratio between phosphorylated and total MEK, and phosphorylated and total ERK1/2 in a. c Downregulation of miR-663b decreased YAP1 and CD44 protein expression in HCT116 and HT29 cells. d Quantification of YAP1 and CD44 expression in c. e Flow cytometry analysis of CD44-positive cells in HCT116 cells transfected with miR-NC inhibitor or miR-663b inhibitor. f Flow cytometry analysis of CD44-positive cells in HT29 cells transfected with miR-NC inhibitor or miR-663b inhibitor. *p < 0.05; **p < 0.01; ***p < 0.001. All experiments were repeated three times
TNK1 was a direct target gene of miR-663b in colorectal cancer cells
We performed bioinformatic analysis to predict potential target genes of miR-663b. We selected TNK1 for further validation based on the knowledge that TNK1 was a confirmed tumor suppressor of colorectal cancer via the inactivation of Ras/Raf signaling. As shown in Fig. 4a, miR-663b could be complementary base paired to 3′UTR of TNK1 mRNA. In both HCT116 and HT29 cells, miR-663b downregulation increased TNK1 mRNA expression (Fig. 4b). Moreover, western blotting further showed that miR-663b downregulation elevated TNK1 protein expression in HCT116 and HT29 cells (Fig. 4c, d). To investigate whether miR-663b directly regulated TNK1 expression, dual luciferase assay was performed. Transfection of miR-663b mimic increased miR-663b expression in HCT116 and HT29 cells (Fig. 4e). It was observed that overexpression of miR-663b decreased relative luciferase activity of TNK1 3′UTR-wild type in HCT116 cells (Fig. 4f). Similarly, overexpression of miR-663b decreased relative luciferase activity of TNK1 3′UTR-wild type in HT29 cells (Fig. 4g). These data indicated that miR-663b directly repressed TNK1 expression in colorectal cancer cells.

MiR-663b directly target 3′UTR of TNK1 mRNA. a 3′UTR of TNK1 mRNA included binding sites for miR-663b. b RT-qPCR showed that downregulation of miR-663b elevated TNK1 mRNA in HCT116 and HT29 cells. c Western blotting showed that downregulation of miR-663b elevated TNK1 protein expression in HCT116 and HT29 cells. d Quantification of TNK1 protein expression in c. e Transfection of miR-663b mimic elevated miR-663b expression in HCT116 and HT29 cells. f In the dual luciferase reporter assay, miR-663b overexpression decreased relative luciferase activity of TNK1 3′UTR-wild type in HCT116 cells. g In the dual luciferase reporter assay, miR-663b overexpression decreased relative luciferase activity of TNK1 3′UTR-wild type in HT29 cells. **p < 0.01; ***p < 0.001. All experiments were repeated three times
MiR-663b regulated YAP1 signaling via repression of TNK1
The regulatory association among miR-663b, TNK1 and Ras/Raf signaling indicated that miR-663b might regulate Ras/Raf signaling via TNK1. We silenced TNK1 in HCT116 cells via transfection of TNK1 siRNA (Fig. 5a, b). Furthermore, miR-663b downregulation elevated TNK1 and decreased YAP1 and CD44 protein expression while TNK1 silencing attenuated elevated TNK1 and downregulation of YAP1 and CD44 expression (Fig. 5c, d). The flow cytometry analysis indicated that the percentage of CD44 positive cells was decreased after miR-663b downregulation which was attenuated towards TNK1 silencing (Fig. 5e).

TNK1 was involved in regulation of YAP1 signaling by miR-663b. a Transfection of TNK1 siRNA decreased TNK1 protein expression in HCT116 cells. b Quantitative analysis of TNK1 expression in a. c Western blotting showed that miR-663b downregulation elevated TNK1, decreased YAP1 and CD44 protein expression which was reversed after TNK1 silencing in HCT116 cells. d Quantification of TNK1, YAP1 and CD44 protein expression in c. e The flow cytometry was used to detect CD44 expression on the surface of HCT116 cells. Downregulation of miR-663b decreased proportion of CD44 positive cells and TNK1 silencing elevated proportion of CD44 positive cells. **p < 0.01; ***p < 0.001. All experiments were repeated three times
MiR-663b regulated cell proliferation and sphere forming via repression of TNK1
To study the involvement of TNK1 in the function of miR-663b, we detected cell viability in HCT116 cells treated with miR-663b inhibitor with or without TNK1 silencing. Downregulation of miR-663b decreased cell viability and silencing of TNK1 elevated cell viability (Fig. 6a). Similarly, TNK1 silencing also attenuated reduction of cell viability induced by miR-663b downregulation in HT29 cells (Fig. 6b). The sphere-forming ability of HCT116 was inhibited by miR-663b inhibitor and reversed after TNK1 silencing in HCT116 and HT29 cells (Fig. 6c, d). The data above demonstrated that miR-663b controls cell proliferation and stemness via targeting TNK1 in colorectal cancer cells.

Downregulation of miR-663b inhibited cell proliferation and sphere forming via elevation of TNK1. a Downregulation of miR-663b inhibited cell proliferation which was reversed after TNK1 silencing in HCT116 cells. b Downregulation of miR-663b inhibited cell proliferation which was reversed after TNK1 silencing in HT29 cells. c Downregulation of miR-663b inhibited sphere forming which was reversed after TNK1 silencing in HCT116 cells. d Downregulation of miR-663b inhibited sphere forming which was reversed after TNK1 silencing in HT29 cells. **p < 0.01; ***p < 0.001. All experiments were repeated three times
TNK1 was reversely correlated with miR-663b expression in colorectal tumors and normal tissues
To investigate the clinical relevance of miR-663b/TNK1, we detected TNK1 expression in 48 pairs of tumors and normal tissues from 48 patients with colorectal cancer. RT-qPCR showed that TNK1 mRNA expression was greatly decreased in tumors (Fig. 7a). We also performed western blotting to detect TNK1 expression in 12 pairs of tumors and normal tissues. TNK1 protein expression was significantly decreased in colorectal tumors (Fig. 7b, c). Moreover, Pearson correlation analysis indicated a strong negative correlation between miR-663b and TNK1 expression in tumors and normal tissues from those patients (Fig. 7d). These results suggested a potential role of miR-663b/TNK1 during colorectal cancer development.

Expression of TNK1 was reversely correlated with miR-663b in tumors and normal tissues. a RT-qPCR was performed to detect TNK1 mRNA expression in 48 pairs of colorectal tumors and normal tissues. The data suggested that TNK1 mRNA was decreased in colorectal tumors. b Representative western blotting showed that TNK1 was downregulated in colorectal tumors compared to normal tissues from two colorectal cancer patients. c TNK1 protein expression was significantly decreased in 12 colorectal tumors compared to their counterparts. d Pearson correlation analysis showed that the TNK1 mRNA expression was negatively correlated with miR-663b levels in tumors and normal tissues from 48 patients with colorectal cancer. ***p < 0.001. All experiments were repeated three times
Discussion
The published data suggested a controversial role of miR-663b in cancer cells. For instance, the increased expression of miR-663 was associated with good prognosis of breast cancer patients [26]. MiR-663 was downregulated in pancreatic cancer and inhibited cancer cell proliferation and invasion via targeting eEF1A2 [27]. MiR-663 suppressed glioblastoma progression via targeting oncogene CXCR4 [28]. In hepatocellular cancer, miR-663 was involved in endoplasmic reticulum stress induced cell apoptosis and inhibited cell proliferation [29], which supported a tumor suppressor role of miR-663 in cancer cells. However, there were studies about the oncogenic role of miR-633 in cancer cells, for instance, miR-663 directly controlled two targets PUMA/BBC3 and BTG2 to regulate apoptosis and sustained NSCLC cell survival [30]. In nasopharyngeal carcinoma, miR-663b promoted cancer cell proliferation, migration, invasion and epithelial–mesenchymal transition by targeting SMAD7 and TUSC2 [31, 32]. In addition, miR-663b also suppressed bladder cancer via targeting TUSC2 [33]. A most recent study discovered that miR-663 was upregulated in serum from colorectal cancer patients compared with healthy volunteers and could serve as an independent biomarker for colorectal cancer diagnosis [21]. Based on previously released miRNA array data, our bioinformatic analysis identified miR-663b as one of most significantly upregulated miRNAs in colon adenoma and carcinoma. Our RT-qPCR results confirmed the upregulation of miR-663b in colorectal tumors. Kaplan–Meier analysis suggested that high expression of miR-663b was associated with poor prognosis of patients. Cancer stem cells are featured with self-renewal ability and play major roles during colorectal cancer initiation, recurrence and drug resistance [34]. In cell-based assays, we observed that miR-663b downregulation suppressed cell proliferation and stemness of colorectal cancer cells. Our study revealed a pivotal role of miR-663b in colorectal cancer.
TNK1 is a recently identified tumor suppressor [35]. Knockout of TNK1 induced spontaneous cancers in mice in various sites including colon. Via TargetScan, it was found that TNK1 was a potential target gene of miR-663b. Western blotting, RT-qPCR and dual luciferase reporter assay provided evidences that miR-663b directly targeted TNK1 mRNA to downregulate TNK1 expression in colorectal cancer cells. These results indicated TNK1 was a novel target gene of miR-663b and first provided data on how TNK1 was regulated in cancer cells. TNK1 upregulated the expression of Ras-negative regular Rap1A and downregulated the activation of Ras pathway in cancer cells [11]. In two colorectal cancer cell lines, we also observed inactivation of Ras signaling after miR-663b downregulation, in HCT116 cells, silencing of TNK1 reversed miR-663b downregulation-induced inactivation of Ras signaling. These data suggested that miR-663b repressed TNK1 expression to control Ras signaling activity. The stemness of colorectal cancer cells was maintained via Ras signaling-dependent YAP1 expression [24]. The downregulation of YAP1 and CD44, a YAP1 target gene, was observed after miR-663b inhibition in colorectal cancer cells. Furthermore, silencing of TNK1 reversed miR-663b-downregulation-induced downregulation of YAP1 and CD44. These findings implied that miR-663b might control proliferation and stemness of colorectal cancer cells via targeting TNK1. Indeed, we found that TNK1 silencing reversed miR-663b inhibitor-induced cell proliferation and stemness inhibition in two colorectal cancer cell lines. The data supported a miR-663b/TNK1/Ras/YAP1 axis in colorectal cancer.
Our study highlighted a pivotal role of miR-663b in colorectal cancer. The results of cell-based assays showed that miR-663b promoted the expression of colorectal cancer stemness-related genes via targeting TNK1 to activate Ras pathway, provided novel insights into the role of miR-663b and the regulation of TNK1 in colorectal cancer.
References
Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–93.
Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–502.
Siveen KS, Raza A, Ahmed EI, et al. The role of extracellular vesicles as modulators of the tumor microenvironment, metastasis and drug resistance in colorectal cancer. Cancers (Basel). 2019;11:746.
Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–28.
Chen D, Gu K, Wang H. Optimizing sequential treatment with anti-EGFR and VEGF mAb in metastatic colorectal cancer: current results and controversies. Cancer Manag Res. 2019;11:1705–16.
Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351(Pt 2):289–305.
Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65.
Jiang H, Wu L, Chen J, et al. Sulfiredoxin promotes colorectal cancer cell invasion and metastasis through a novel mechanism of enhancing EGFR signaling. Mol Cancer Res. 2015;13:1554–66.
Klampfer L, Swaby LA, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Oncogenic Ras increases sensitivity of colon cancer cells to 5-FU-induced apoptosis. Oncogene. 2005;24:3932–41.
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45.
Hoare K, Hoare S, Smith OM, Kalmaz G, Small D, Stratford May W. Kos1, a nonreceptor tyrosine kinase that suppresses Ras signaling. Oncogene. 2003;22:3562–77.
Hoehn GT, Stokland T, Amin S, et al. Tnk1: a novel intracellular tyrosine kinase gene isolated from human umbilical cord blood CD34+/Lin-/CD38-stem/progenitor cells. Oncogene. 1996;12:903–13.
Hoare S, Hoare K, Reinhard MK, Lee YJ, Oh SP, May WS Jr. Tnk1/Kos1 knockout mice develop spontaneous tumors. Cancer Res. 2008;68:8723–32.
Kavitha N, Vijayarathna S, Jothy SL, et al. MicroRNAs: biogenesis, roles for carcinogenesis and as potential biomarkers for cancer diagnosis and prognosis. Asian Pac J Cancer Prev. 2014;15:7489–97.
Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions–beyond repression of gene expression. Nat Rev Genet. 2014;15:599–612.
Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–62.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Liu F, Di Wang X. miR-150-5p represses TP53 tumor suppressor gene to promote proliferation of colon adenocarcinoma. Sci Rep. 2019;9:6740.
You Q, Li H, Liu Y, et al. MicroRNA-650 targets inhibitor of growth 4 to promote colorectal cancer progression via mitogen activated protein kinase signaling. Oncol Lett. 2018;16:2326–34.
Xie HH, Huan WT, Han JQ, Ren WR, Yang LH. MicroRNA-663 facilitates the growth, migration and invasion of ovarian cancer cell by inhibiting TUSC2. Biol Res. 2019;52:18.
Wang N, Zeng L, Li Z, Zhen Y, Chen H. Serum miR-663 expression and the diagnostic value in colorectal cancer. Artif Cells Nanomed Biotechnol. 2019;47:2650–3.
Lee SA, Kwon HC, Park MA, et al. Impact of the new AJCC staging system and adjuvant treatment in rectal cancer. Cancer Res Treat. 2004;36:121–7.
Mizukami T, Izawa N, Nakajima TE, Sunakawa Y. Targeting EGFR and RAS/RAF signaling in the treatment of metastatic colorectal cancer: from current treatment strategies to future perspectives. Drugs. 2019;79:633–45.
Qian F, Xiao J, Gai L, Zhu J. HMGB1-RAGE signaling facilitates Ras-dependent Yap1 expression to drive colorectal cancer stemness and development. Mol Carcinog. 2019;58:500–10.
Hofmann M, Rudy W, Gunthert U, et al. A link between ras and metastatic behavior of tumor cells: ras induces CD44 promoter activity and leads to low-level expression of metastasis-specific variants of CD44 in CREF cells. Cancer Res. 1993;53:1516–21.
Carden T, Singh B, Mooga V, Bajpai P, Singh KK. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J Biol Chem. 2017;292:20694–706.
Zang W, Wang Y, Wang T, et al. miR-663 attenuates tumor growth and invasiveness by targeting eEF1A2 in pancreatic cancer. Mol Cancer. 2015;14:37.
Shi Y, Chen C, Yu SZ, et al. miR-663 suppresses oncogenic function of CXCR28 in glioblastoma. Clin Cancer Res. 2015;21:4004–13.
Huang Y, Liu J, Fan L, et al. miR-663 overexpression induced by endoplasmic reticulum stress modulates hepatocellular carcinoma cell apoptosis via transforming growth factor beta 1. Onco Targets Ther. 2016;9:1623–33.
Fiori ME, Villanova L, Barbini C, De Angelis ML, De Maria R. miR-663 sustains NSCLC by inhibiting mitochondrial outer membrane permeabilization (MOMP) through PUMA/BBC3 and BTG2. Cell Death Dis. 2018;9:49.
Liang S, Zhang N, Deng Y, et al. miR-663b promotes tumor cell proliferation, migration and invasion in nasopharyngeal carcinoma through targeting TUSC2. Exp Ther Med. 2017;14:1095–103.
Wang M, Jia M, Yuan K. MicroRNA-663b promotes cell proliferation and epithelial mesenchymal transition by directly targeting SMAD7 in nasopharyngeal carcinoma. Exp Ther Med. 2018;16:3129–34.
Chen J, Song B, Kong G. MicroRNA663b downregulation inhibits proliferation and induces apoptosis in bladder cancer cells by targeting TUSC2. Mol Med Rep. 2019;19:3896–902.
Ma L, Wang L, Shan Y, et al. Suppression of cancer stemness by upregulating Ligand-of-Numb protein X1 in colorectal carcinoma. PLoS ONE. 2017;12:e0188665.
May WS, Hoare K, Hoare S, Reinhard MK, Lee YJ, Oh SP. Tnk1/Kos1: a novel tumor suppressor. Trans Am Clin Climatol Assoc. 2010;121:281–92 (discussion 92-3).
Author information
Authors and Affiliations
Contributions
SH, ZY and HW participated in the design and performance of the experiments; SH, ZY, HW, YS and MB contributed to the collection of samples and clinical data; ZY and HW analyzed the data; HW and LD supervised and wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
We declare that we have no conflict of interest in the current study.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of The First Hospital of Jilin University (No. FHJU20120603) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hong, S., Yan, Z., Wang, H. et al. miR-663b promotes colorectal cancer progression by activating Ras/Raf signaling through downregulation of TNK1. Human Cell 33, 104–115 (2020). https://doi.org/10.1007/s13577-019-00294-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-019-00294-w