
Abstract
Long noncoding RNAs (lncRNAs) have been found to be abnormally expressed in cancer, and lncRNA small nucleolar RNA host genes (SNHGs) play critical roles in tumour progression. SNHG3 has been identified as an oncogene in multiple tumour types. However, the role of SNHG3 in breast cancer has not been reported. In this study, we found that SNHG3 was upregulated and associated with tumour malignancy in patients with breast cancer. SNHG3 knockdown inhibited the growth and metastatic capabilities of breast cancer cells in vitro and vivo. We used bioinformatics prediction and functional assay validation to determine that SNHG3 upregulation inhibited miR-384 activity and led to hepatoma-derived growth factor (HDGF) overexpression in breast cancer cells. The findings of this study show that SNHG3 functions as an oncogene in breast cancer and promotes breast cancer cell proliferation and invasion by regulating the miR-384/HDGF axis. The present study might provide a new target for the treatment of breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Breast cancer is one of the most common malignant tumours in women [1, 2]. Despite advances in diagnosis and treatment, the survival rate remains low for breast cancer patients [3]. The abnormal metastasis of breast cancer cells is a major contributor to breast cancer patient death [4]. Therefore, investigating the mechanisms underlying breast cancer progression is important.
LncRNAs are defined as RNAs with a length of more than 200 nucleotides and no protein-coding ability [5, 6]. Previous reports have shown that lncRNAs regulate gene expression at the epigenetic, transcriptional and posttranscriptional levels and participate in various physiological processes by interacting with proteins and nucleic acids [7, 8]. Increasing evidence indicates that aberrant lncRNA expression is involved in the occurrence, development and metastasis of tumours [9,10,11]. In breast cancer, some lncRNAs have been identified as oncogenes or tumour suppressors and have potential as diagnostic or prognostic markers for breast cancer patients [12, 13].
The potential function and mechanism of the lncRNA SNHG3 in breast cancer remain unclear. In a previous study, Zhang et al. reported that SNHG3 correlates with malignant status and poor prognosis in hepatocellular carcinoma [14]. Hong et al. reported that the expression of SNHG3 is associated with poor prognosis and can promote the malignant progression of ovarian cancer [15]. In this study, we found that SNHG3 expression was significantly higher in breast cancer tissues than in adjacent normal tissues. Moreover, high SNHG3 expression levels were associated with tumour malignancy in breast cancer patients. Then we examined the biological functions of SNHG3 in breast cancer cell proliferation and invasion using a series of experiments. Recent studies have shown that lncRNAs act as competing endogenous RNAs (ceRNAs) to sponge miRNAs and participate in physiological and pathological processes [16]. When investigating whether SNHG3 interacts with miRNAs, we found that SNHG3 possesses a potential binding site for miR-384 using the starBase web tool. Although the roles of miR-384 in many types of cancers have been investigated previously [17, 18], the effect of miR-384 on breast cancer has rarely been reported. In this study, we showed that SNHG3 interacted with miR-384 and inhibited its expression. Moreover, we showed that SNHG3 promoted breast cancer cell proliferation and invasion by regulating the miR-384/HDGF axis. In conclusion, our data indicate that SNHG3 has potential clinical value and can be used as a target for the treatment of breast cancer.
Materials and methods
Clinical specimens
The 60 breast cancer and paired adjacent normal tissues used in this study were obtained from Zibo Central Hospital (Zibo, Shandong, China). The breast cancer conditions were diagnosed by two pathologists following the American Society of Clinical Oncology guidelines [19]. The tissue samples obtained upon resection were stored in liquid nitrogen immediately until further use. The clinical characteristics of the patients are listed in Table 1. All patients in this study provided signed informed consent, and the study was approved by the Ethics Committee of Zibo Central Hospital.
Cell culture and transfection
The human breast cancer cell lines MDA-MB-231 and MCF-7 and the immortalized breast epithelial cell line MCF-10A were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% foetal bovine serum (FBS, Gibco-BRL, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Beyotime Institute of Biotechnology, Shanghai, China). All the cells were maintained in humidified air at 37 °C with 5% CO2. SNHG3 lentivirus particles (si-SNHG3), control lentivirus particles (si-NC), shRNA expression lentiviral plasmid (sh-HDGF) and control lentiviral plasmid (sh-NC) were purchased from GenePharma Biotechnology (Shanghai, China). Lentivirus infection was performed according to the manufacturer’s instructions. Cells at 70% confluence were treated with 5 μg/mL polybrene (GenePharma Biotechnology) before virus infection, and the cells were transferred to fresh medium after 1 day. Cells with stable expression were selected using medium containing puromycin (Sigma, USA). miR-384 mimic, miR-384 inhibitor and the corresponding negative controls were purchased from GenePharma Biotechnology. Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) was used to perform miR-384 mimic, miR-384 inhibitor and corresponding negative control transfections according to the manufacturer’s instructions. All sequences are listed in Supplementary Table S1.
Total RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from tissues and cells using RNAiso Plus (TAKARA Biotechnology, Dalian, China) according to the manufacturer’s instructions. For SNHG3, miR-384 and HDGF mRNA expression, qRT-PCR was performed using a SYBR Green-based PCR Kit (TaKaRa Biotechnology, Dalian, China). The expression level of each gene is presented as relative value to that of the internal controls β-actin and U6 snRNA. All of the primers used are listed in Table 2. The experiments were performed independently in triplicate, and the data are presented as the mean ± standard error of the mean (SEM).
Cell proliferation assay
A Cell Counting Kit-8 (CCK-8, Beyotime Institute of Biotechnology, Shanghai, China) assay was used to measure the proliferation of breast cancer cells according to the manufacturer’s instructions. Briefly, cells (4 × 103 per well) were seeded into a 96-well plate and cultured at 24-h intervals for 3 days. Subsequently, the cells were treated with 10% CCK-8 solution and incubated at 37 °C for 2 h. The absorbance at 450 nm was measured with a microplate reader (Thermo Fisher, USA). The experiments were performed independently in triplicate, and the data are presented as the mean ± SEM.
Colony formation assay
Cells were seeded into a six-well plate at a density of 0.2 × 103 cells/well and cultured for 10 days at 37 °C in a 5% CO2 atmosphere. The plate was then fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for 20 min. The plate was washed twice with PBS, and the numbers of colonies with more than 50 cells were counted and photographed. The experiment was performed independently in triplicate, and the data are presented as the mean ± SEM.
Cell migration and invasion assays
First, breast cancer cells were starved in DMEM without FBS for 12 h. Then 6 × 104 cells resuspended in 0.2 ml serum-free DMEM were added to the upper uncoated (for migration) or Matrigel matrix-coated (for invasion, BD, CA, USA) chambers, and DMEM containing 20% FBS was added to the lower chambers as a chemoattractant at 37 °C in humidified 5% CO2. Twelve hours later, the nonmigrated or noninvaded cells were wiped away. The cells that migrated or invaded to the lower chamber were fixed and stained with a crystal violet solution. Three low magnification areas (× 100) were selected randomly, and the number of migrated or invaded cells was counted. The experiment was performed independently in triplicate, and the data are presented as the mean ± SEM.
Animal experiments
Four-week-old male BALB/c nude mice were purchased from the Shanghai Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China) and divided randomly into two groups (n = 4 per group). Then 1 × 107 breast cancer cells were injected subcutaneously into the flank of each nude mouse. Tumour volumes were measured at indicated time points. Tumour weight was determined 4 weeks after injection. In the tail vein injection experiment, MDA-MB-231 cells transfected with si-SNHG3 or negative control were injected into the tail vein of each mouse. Eight weeks after injection, the lungs of the mice were excised, and visible tumour nodes on the lung surface were counted. The animal experiment was approved by the Ethics Committee of Zibo Central Hospital.
Luciferase reporter assay
Sequences containing wild-type (WT) or mutant (MUT) SNHG3-binding sites were subcloned into the pGL3-control vector (Promega, Madison, WI, USA). Putative miR-384 binding sites in the 3′ untranslated region (UTR) of wild-type HDGF (WT-HDGF) or mutant HDGF (MUT-HDGF) mRNA were synthesized and inserted into the pGL3-control vector. For the reporter assay, breast cancer cells were plated onto 24-well plates and transfected with WT-SNHG3 or MUT-SNHG3, WT-HDGF or MUT-HDGF and miR-384 mimic using Lipofectamine 2000. After transfection for 48 h, the cells were harvested and assayed with a luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The experiment was performed independently in triplicate, and the data are presented as the mean ± SEM.
RNA immunoprecipitation (RIP) assay
RIP assays were performed according to the instructions of the EZ-Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA). Cells in different groups were lysed in lysis buffer containing protease inhibitor cocktail and RNase inhibitor. Cells were incubated with RIP buffer containing magnetic beads coated with Ago2 antibodies (Millipore). IgG was used as a negative control. After incubation for 2 h at 4 °C, the coprecipitated RNA was eluted from the beads and measured by PCR analysis. The experiment was performed independently in triplicate, and the data are presented as the mean ± SEM.
Western blot analysis
Cells were collected and lysed in ice-cold RIPA buffer containing protease inhibitor (Beyotime Institute of Biotechnology). Proteins were separated using sodium dodecyl sulphate–polyacrylamide gel electrophoresis and then electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, USA). Following blocking with 5% skim milk solution, membranes were incubated at 4 °C overnight with specific antibodies against HDGF (1:1000) and β-actin (1:2000) (all purchased from Proteintech Group, Rosemont, IL, USA). Subsequently, the membranes were incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody (Proteintech) at room temperature for 1 h, followed by enhanced chemiluminescence detection (Beyotime Institute of Biotechnology) and autoradiography. The experiment was performed independently in triplicate.
Statistical analysis
Statistical analyses were performed using GraphPad software (GraphPad Prism version 5.0, San Diego, USA) and SPSS software (version 17.0; SPSS, Inc., Chicago, IL, USA). The results are expressed as mean ± SEM from at least three independent experiments. A two-tailed paired Student’s t test was conducted for the analysis of two groups. A Mann–Whitney U test was carried out to analyse the relationship between SNHG3 expression and the clinicopathological features of the breast cancer patients. P < 0.05 was considered to indicate a statistically significant difference. Statistically significant data are indicated by asterisks (*P < 0.05, **P < 0.01 and ***P < 0.01) in the figures.
Results
SNHG3 was upregulated and associated with tumour malignancy in breast cancer
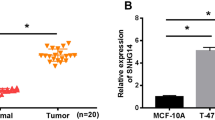
First, we examined SNHG3 expression in 60 pairs of breast cancer tissues. qRT-PCR showed that SNHG3 expression was significantly higher in breast cancer tissues than in adjacent normal tissues (Fig. 1a). Moreover, we investigated the correlation of SNHG3 expression with various clinicopathological parameters of breast cancer patients. The statistical analyses demonstrated that SNHG3 expression was significantly associated with histological grade, lymph node metastasis, advanced tumour–node–metastasis (TNM) stage, and oestrogen receptor (ER) and human epidermal growth factor receptor 2 (Her-2) status (Fig. 1b, c and Table 1). In addition, qRT-PCR showed that SNHG3 expression was higher in breast cancer cell lines (MDA-MB-231 and MCF-7) than in MCF-10A cells (Fig. 1d). Together, these data indicate that SNHG3 is upregulated and associated with tumour malignancy in breast cancer specimens.

SNHG3 is upregulated in breast cancer. a qRT-PCR was used to examine SNHG3 expression in breast cancer tissue (BCT) and adjacent normal tissue (ANT). b SNHG3 expression was higher in breast cancer patients with lymph node metastasis (LM) than in patients with no lymph node metastasis (NLM). c SNHG3 expression was higher in breast cancer patients with TNM stage II + III than in those with TNM stage I. d qRT-PCR was used to examine SNHG3 expression in breast cancer cell lines and immortalized breast epithelial cells. The experiments were performed independently in triplicate, and the data are presented as the mean ± SEM, ***P < 0.001
SNHG3 promoted breast cancer cell proliferation and invasion in vitro and in vivo
To explore the roles of SNHG3 in breast cancer tumourigenesis, MDA-MB-231 and MCF-7 cells were transfected with si-SNHG3 and si-NC (Fig. 2a). The CCK-8 and colony formation assay results indicated that SNHG3 inhibition significantly reduced breast cancer cell proliferation and colony formation abilities (Fig. 2b, c). Transwell assays revealed that SNHG3 suppression significantly reduced breast cancer cell migration and invasion abilities (Fig. 2d). The same effects are shown in Supplementary Fig. S1 for cells transfected with si-SNHG3-1. Next, we further explored the function of SNHG3 in vivo. Stable MDA-MB-231 cells transfected with si-SNHG3 and si-NC were injected subcutaneously into nude mice. Continuous measurements of tumour growth were taken every 4 days, followed by tumour excision and weighing 4 weeks after injection. si-SNHG3 significantly suppressed the average tumour volume and weight compared with si-NC (Fig. 2e, f). Furthermore, a tail vein injection experiment was adopted to explore the effect of SNHG3 knockdown on breast cancer cell metastasis in vivo. The results revealed that si-SNHG3 significantly reduced the number of lung metastatic nodules compared with si-NC (Fig. 2g). These results indicate that SNHG3 might play critical roles in breast cancer progression.

SNHG3 knockdown inhibits breast cancer cell proliferation, migration and invasion in vitro and in vivo. a qRT-PCR verified the interference efficiency of SNHG3 in breast cancer cell lines. b A CCK-8 assay was performed to measure breast cancer cell proliferation after transfection with si-SNHG3 and si-NC. c A colony formation assay was performed to measure breast cancer cell proliferation after transfection. d Transwell assays were performed to determine the effects of SNHG3 knockdown on breast cancer cell migration and invasion. e Tumour size was measured every 4 days. The tumour volume was smaller in the si-SNHG3 group than in the si-NC group. f The xenograft tumour weight was significantly lower in the si-SNHG3 group than in the si-NC group. g The number of visible metastatic nodules on the lung surface was counted. The experiments were performed independently in triplicate, and the data are presented as the mean ± SEM, ***P < 0.001
SNHG3 interacts with miR-384 and decreases its expression in breast cancer
To further explore the underlying molecular mechanism by which SNHG3 regulates breast cancer, we performed a bioinformatics prediction analysis and found that miR-384 was a potential candidate. Bioinformatics analysis showed that SNHG3 might interact with miR-384 (Fig. 3a). A dual luciferase reporter assay showed that miR-384 mimic significantly reduced the luciferase activity of WT-SNHG3 in breast cancer cells (Fig. 3b). qRT-PCR showed that SNHG3 knockdown remarkably increased miR-384 expression in breast cancer cells (Fig. 3c). In addition, miR-384 expression was significantly decreased in breast cancer tissues and cell lines (Fig. 3d, e). A correlation analysis revealed that SNHG3 expression was negatively correlated with miR-384 expression in breast cancer tissues (Fig. 3f). Through starBase v2.0 Program prediction, 16 miRNAs were predicted as possible targets of SNHG3 (Supplementary Table S2). Among the 16 candidate miRNAs predicted by starBase, we screened and analysed miR-758-3p and miR-384, whose effects on breast cancer have been rarely reported. Next, we identified functional miRNAs that may interact with SNHG3 in breast cancer cells. RIP assays showed that SNHG3 and miR-384 (not miR-758-3p) expression was significantly enriched in the Ago2 pellet compared to that in the IgG control pellet (Fig. 3g). These data indicate that SNHG3 interacts with miR-384 and decreases its expression in breast cancer.

SNHG3 interacts with miR-384 and decreases its expression in breast cancer. a Predicted position of the miR-384-binding sites in SNHG3. b A dual luciferase reporter assay showed that miR-384 mimic significantly decreased the luciferase activity of WT-SNHG3 in breast cancer cells. c SNHG3 inhibition of miR-384 expression in breast cells. d, e The expression level of miR-384 in BCT and cell lines was detected by qRT-PCR. f SNHG3 expression was negatively correlated with miR-384 expression in BCT. g RIP assay showing that SNHG3 and miR-384 were both enriched in Ago2-containing miRNAs. The experiments were performed independently in triplicate, and the data are presented as the mean ± SEM, ***P < 0.001
miR-384 inhibits breast cancer cell proliferation and invasion by targeting HDGF
To confirm the function of miR-384, we transfected miR-384 mimic or NC mimic sequences into breast cancer cells. The CCK-8 and colony formation assay results indicated that miR-384 significantly reduced breast cancer cell proliferation and colony formation abilities (Fig. 4a, b). Transwell assays revealed that miR-384 significantly reduced breast cancer cell migration and invasion abilities (Fig. 4c). Bioinformatics analyses (TargetScan) revealed that HDGF is a potential target of miR-384 (Fig. 4d). To confirm HDGF as a direct target of miR-384, we engineered luciferase reporter constructs containing the WT or MUT 3′ UTR of the HDGF gene. The luciferase reporter assay showed that miR-384 significantly decreased the luciferase activity of the WT-HDGF 3′ UTR but not that of the MUT-HDGF 3′ UTR in MDA-MB-231 and MCF-7 cells (Fig. 4e). Furthermore, qRT-PCR and western blot analysis revealed that miR-384 mimic transfection in MDA-MB-231 and MCF-7 cells led to a considerable reduction in both the mRNA and protein levels of HDGF (Fig. 4f, g). We then detected HDGF expression in breast cancer and adjacent nontumour tissues by qRT-PCR. HDGF expression was obviously higher in breast cancer tissues than in adjacent nontumour tissues (Fig. 4h). Thereafter, a correlation analysis indicated that the miR-384 expression level was inversely correlated with the HDGF mRNA expression level in breast cancer tissues (Fig. 4i). Taken together, these data show that HDGF is a direct target of miR-384 in breast cancer.

HDGF is a functional downstream target of miR-384. a The proliferation of breast cancer cells transfected with miR-384 mimic or NC mimic was determined by CCK-8 assay. b A colony formation assay showed that miR-384 inhibited breast cancer cell viability. c Transwell assays detected the migration and invasion abilities of breast cancer cells transfected with miR-384 mimic. d Bioinformatics analysis revealed the predicted binding sites between HDGF and miR-384. e A dual luciferase reporter assay demonstrated that miR-384 mimic significantly decreased the luciferase activity of WT-HDGF in breast cancer cells. f qRT-PCR detected HDGF mRNA expression levels after miR-384 mimic transfection in breast cancer cells. g The protein levels of HDGF were decreased in breast cancer cells transfected with miR-384 mimic. h HDGF expression was significantly higher in BCT than in ANT according to qRT-PCR analysis. i A correlation analysis revealed a negative relationship between HDGF and miR-384 expression in BCT. The experiments were performed independently in triplicate, and the data are presented as the mean ± SEM, ***P < 0.001
SNHG3 promotes cell proliferation and invasion via the miR-384/HDGF axis
To test whether SNHG3 can regulate HDGF via miR-384 in breast cancer cells, a western blot assay was performed. Transfection with si-SNHG3 significantly decreased HDGF expression. Transfection with miR-384 inhibitor significantly increased HDGF expression, and cotransfection with si-SNHG3 and miR-384 inhibitor increased HDGF expression compared with cotransfection with si-SNHG3 and NC inhibitor (Fig. 5a). Next, a correlation analysis revealed that HDGF expression was positively correlated with SNHG3 expression in breast cancer tissues (Fig. 5b). Furthermore, CCK-8, colony formation and transwell assays showed that the miR-384 inhibitor significantly rescued the proliferation, colony formation, migration and invasion abilities induced by si-SNHG3 in breast cancer cells (Fig. 5c–e). In addition, we knocked down HDGF expression in breast cancer cells using shRNA (Fig. 5f). The results show that HDGF knockdown cancelled the effect of miR-384 downregulation with a miR-384 inhibitor. Compared to those in cells cotransfected with miR-384 inhibitor and sh-NC, cell proliferation, colony formation, migration, and invasion were significantly suppressed in cells cotransfected with miR-384 inhibitor and sh-HDGF (Fig. 5g–i). These results indicate that SNHG3 promotes breast cancer progression by regulating the miR-384/HDGF axis (Fig. 5j).

SNHG3 promotes cell proliferation and invasion via the HDGF/miR-384 axis. a SNHG3 controls HDGF expression by regulating miR-384 in breast cancer cells, as analysed by western blotting. b SNHG3 expression was positively correlated with HDGF expression in BCT. c A CCK-8 assay showed that miR-384 inhibitor rescued the proliferation effects of SNHG3 knockdown on breast cancer cells. d A colony formation assay showed that miR-384 inhibitor significantly rescued the viability effects of SNHG3 knockdown on breast cancer cells. e Transwell assays showed that miR-384 inhibitor significantly rescued the migration and invasion effects of SNHG3 knockdown on breast cancer cells. f Western blot analysis of the effects of sh-HDGF on HDGF expression. g CCK-8 assay to investigate breast cancer cell proliferation after cotransfection with miR-384 inhibitor and the sh-HDGF. h Colony formation assay to determine the colony number of breast cancer cells after cotransfection with miR-384 inhibitor and the sh-HDGF. i Transwell assay to investigate breast cancer cell migration and invasion after cotransfection with miR-384 inhibitor and the sh-HDGF. j Schematic diagram of the mechanism derived from the experimental results of this study. The experiments were performed independently in triplicate, and the data are presented as the mean ± SEM, ***P < 0.001
Discussion
Although advancements in clinical therapies have improved clinical conditions, breast cancer patients with an advanced stage and distant metastasis still have a very poor prognosis; therefore, new targets against breast cancer cell proliferation and metastasis are urgently needed. The aberrant expression of lncRNAs is common in human solid tumours and critical for tumour progression [20, 21]. Cao et al. showed that the upregulation of the lncRNA SNHG16 correlated with tumour progression and poor prognosis in bladder cancer [22]. Gao et al. found that the lncRNA SBF2-AS1 regulated cervical cancer progression by targeting the miR-361-5p/FOXM1 signalling pathway [23]. Increasing evidence has indicated that lncRNAs are vital to the development and prognosis of breast cancer [24]. For example, the lncRNA Z38 is highly expressed, and silencing Z38 significantly inhibited cell proliferation and tumourigenesis in breast cancer [25]. The lncRNA FBXL19-AS1 is upregulated in breast cancer, and FBXL19-AS1 promotes cell proliferation and invasion in breast cancer [26].
Among lncRNAs, SNHG3 has been proven to be highly associated with human cancers, including osteosarcoma [27], hepatocellular carcinoma [28], ovarian cancer [15], and colorectal cancer [29]. However, the roles and underlying mechanisms of SNHG3 in breast cancer remain largely unknown. In our study, we demonstrated that the lncRNA SNHG3 was significantly increased in not only breast cancer patient samples but also breast cancer cell lines. High levels of SNHG3 expression were associated with tumour malignancy in breast cancer patients. Functional assays revealed that the downregulation of SNHG3 significantly inhibited the proliferation, colony formation, migration and invasion of breast cancer cells. These results suggest that SNHG3 acts as an oncogenic lncRNA in breast cancer progression.
MicroRNAs (miRNAs) are noncoding RNAs that can regulate a set of target genes through translational repression or mRNA degradation [30]. Recently, increasing studies have shown that lncRNAs act as ‘sponges’ to bind with specific miRNAs, thus regulating multiple diseases [31]. For example, Zhang et al. found that the lncRNA LINC00460 promoted colorectal cancer cell metastasis via sponging miR-939-5p [32]. Du et al. revealed that the lncRNA MAGI2-AS3 inhibits the migration and invasion of breast cancer by sponging miR-374a to upregulate PTEN [33]. Previous studies have shown that miR-384 promotes proliferation and metastasis in many malignant tumours, such as thyroid cancer, pancreatic cancer, hepatocellular carcinoma and colorectal cancer [34,35,36,37]. In this study, luciferase reporter and RIP assays showed that SNHG3 could bind to miR-384 in breast cancer cells. Previous studies have shown that miR-384 might serve as a tumour suppressor in some human tumours [38, 39]. In the present study, we found that miR-384 was significantly decreased and inversely associated with SNHG3 expression in breast cancer tissues. Functional assays showed that miR-384 reduced breast cancer cell viability and invasion and that a miR-384 inhibitor abolished the effects of SNHG3 knockdown on breast cancer cells. In this way, SNHG3 promoted breast cancer progression through the inhibition of miR-384.
Next, we used online biological software to predict that HDGF is a potential target gene of miR-384. HDGF is a heparin-binding growth factor that was first purified from culture media conditioned with the human hepatoma cell line Huh7 [40, 41]. Increasing evidence suggests an important role for HDGF in the progression of many tumours. High HDGF expression correlates with a poor prognosis in breast cancer and promotes cell growth, migration and invasion [42, 43]. Here, we found that knocking down SNHG3 significantly decreased HDGF expression at the mRNA and protein levels in breast cancer cells. Moreover, si-SNHG3 and miR-384 inhibitor cotransfection increased HDGF expression compared with si-SNHG3 and NC inhibitor cotransfection in breast cancer cells. Furthermore, CCK-8, colony formation and transwell assays showed that si-SNHG3 and miR-384 inhibitor cotransfection significantly increased cell proliferation, colony formation, migration and invasion compared with si-SNHG3 and NC inhibitor cotransfection in breast cancer cells. These results suggest that SNHG3 exerts its oncogenic function, at least in part, by regulating the miR-384/HDGF axis.
In conclusion, our studies demonstrated that the lncRNA SNHG3 is highly expressed in breast cancer and promotes proliferation and invasion via the miR-384/HDGF axis in breast cancer cells. These findings provide a novel mechanism for the occurrence and development of breast cancer.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. https://doi.org/10.3322/caac.21387.
Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: epidemiology and etiology. Cell Biochem Biophys. 2015;72(2):333–8. https://doi.org/10.1007/s12013-014-0459-6.
Chou J, Wang B, Zheng T, Li X, Zheng L, Hu J, et al. MALAT1 induced migration and invasion of human breast cancer cells by competitively binding miR-1 with cdc42. Biochem Biophys Res Commun. 2016;472(1):262–9. https://doi.org/10.1016/j.bbrc.2016.02.102.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18(1):5–18. https://doi.org/10.1038/nrc.2017.99.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. https://doi.org/10.1038/nrg.2015.10.
Martens-Uzunova ES, Bottcher R, Croce CM, Jenster G, Visakorpi T, Calin GA. Long noncoding RNA in prostate, bladder, and kidney cancer. Eur Urol. 2014;65(6):1140–51. https://doi.org/10.1016/j.eururo.2013.12.003.
Zhang A, Xu M, Mo YY. Role of the lncRNA-p53 regulatory network in cancer. J Mol Cell Biol. 2014;6(3):181–91. https://doi.org/10.1093/jmcb/mju013.
Cheng J, Chen J, Zhang X, Mei H, Wang F, Cai Z. Overexpression of CRNDE promotes the progression of bladder cancer. Biomed Pharmacother. 2018;99:638–44. https://doi.org/10.1016/j.biopha.2017.12.055.
Sun MD, Zheng YQ, Wang LP, Zhao HT, Yang S. Long noncoding RNA UCA1 promotes cell proliferation, migration and invasion of human leukemia cells via sponging miR-126. Eur Rev Med Pharmacol Sci. 2018;22(8):2233–45. https://doi.org/10.26355/eurrev_201804_14809.
Zhu Y, Qiao L, Zhou Y, Ma N, Wang C, Zhou J. Long non-coding RNA FOXD2-AS1 contributes to colorectal cancer proliferation through its interaction with microRNA-185-5p. Cancer Sci. 2018;109(7):2235–42. https://doi.org/10.1111/cas.13632.
Gu J, Wang Y, Wang X, Zhou D, Zhou M, He Z. Effect of the LncRNA GAS5-MiR-23a-ATG3 axis in regulating autophagy in patients with breast cancer. Cell Physiol Biochem. 2018;48(1):194–207. https://doi.org/10.1159/000491718.
Zhang Y, Li J, Jia S, Wang Y, Kang Y, Zhang W. Down-regulation of lncRNA-ATB inhibits epithelial-mesenchymal transition of breast cancer cells by increasing miR-141-3p expression. Biochem Cell Biol. 2019;97(2):193–200. https://doi.org/10.1139/bcb-2018-0168.
Zhang T, Cao C, Wu D, Liu L. SNHG3 correlates with malignant status and poor prognosis in hepatocellular carcinoma. Tumour Biol. 2016;37(2):2379–85. https://doi.org/10.1007/s13277-015-4052-4.
Hong L, Chen W, Wu D, Wang Y. Upregulation of SNHG3 expression associated with poor prognosis and enhances malignant progression of ovarian cancer. Cancer Biomark. 2018;22(3):367–74. https://doi.org/10.3233/CBM-170710.
Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17(5):272–83. https://doi.org/10.1038/nrg.2016.20.
Song H, Rao Y, Zhang G, Kong X. MicroRNA-384 inhibits the growth and invasion of renal cell carcinoma cells by targeting astrocyte elevated gene 1. Oncol Res. 2018;26(3):457–66. https://doi.org/10.3727/096504017X15035025554553.
Fan N, Zhang J, Cheng C, Zhang X, Feng J, Kong R. MicroRNA-384 represses the growth and invasion of non-small-cell lung cancer by targeting astrocyte elevated gene-1/Wnt signaling. Biomed Pharmacother. 2017;95:1331–7. https://doi.org/10.1016/j.biopha.2017.08.143.
Lyman GH, Somerfield MR, Bosserman LD, Perkins CL, Weaver DL, Giuliano AE. Sentinel lymph node biopsy for patients with early-stage breast cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2017;35(5):561–4. https://doi.org/10.1200/JCO.2016.71.0947.
Jin Y, Feng SJ, Qiu S, Shao N, Zheng JH. LncRNA MALAT1 promotes proliferation and metastasis in epithelial ovarian cancer via the PI3K-AKT pathway. Eur Rev Med Pharmacol Sci. 2017;21(14):3176–84.
Yang Y, Yang H, Xu M, Zhang H, Sun M, Mu P, et al. Long non-coding RNA (lncRNA) MAGI2-AS3 inhibits breast cancer cell growth by targeting the Fas/FasL signalling pathway. Hum Cell. 2018;31(3):232–41. https://doi.org/10.1007/s13577-018-0206-1.
Cao X, Xu J, Yue D. LncRNA-SNHG16 predicts poor prognosis and promotes tumor proliferation through epigenetically silencing p21 in bladder cancer. Cancer Gene Ther. 2018;25(1–2):10–7. https://doi.org/10.1038/s41417-017-0006-x.
Gao F, Feng J, Yao H, Li Y, Xi J, Yang J. LncRNA SBF2-AS1 promotes the progression of cervical cancer by regulating miR-361-5p/FOXM1 axis. Artif Cells Nanomed Biotechnol. 2019;47(1):776–82. https://doi.org/10.1080/21691401.2019.1577883.
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–61. https://doi.org/10.1038/nm.3981.
Deng R, Liu B, Wang Y, Yan F, Hu S, Wang H, et al. High expression of the newly found long noncoding RNA Z38 promotes cell proliferation and oncogenic activity in breast cancer. J Cancer. 2016;7(5):576–86. https://doi.org/10.7150/jca.13117.
Ding Z, Ye P, Yang X, Cai H. LncRNA FBXL19-AS1 promotes breast cancer cells proliferation and invasion via acting as a molecular sponge to miR-718. Biosci Rep. 2019. https://doi.org/10.1042/BSR20182018.
Zheng S, Jiang F, Ge D, Tang J, Chen H, Yang J, et al. LncRNA SNHG3/miRNA-151a-3p/RAB22A axis regulates invasion and migration of osteosarcoma. Biomed Pharmacother. 2019;112:108695. https://doi.org/10.1016/j.biopha.2019.108695.
Zhang PF, Wang F, Wu J, Wu Y, Huang W, Liu D, et al. LncRNA SNHG3 induces EMT and sorafenib resistance by modulating the miR-128/CD151 pathway in hepatocellular carcinoma. J Cell Physiol. 2019;234(3):2788–94. https://doi.org/10.1002/jcp.27095.
Huang W, Tian Y, Dong S, Cha Y, Li J, Guo X, et al. The long non-coding RNA SNHG3 functions as a competing endogenous RNA to promote malignant development of colorectal cancer. Oncol Rep. 2017;38(3):1402–10. https://doi.org/10.3892/or.2017.5837.
Chen E, Xu X, Liu R, Liu T. Small but heavy role: microRNAs in hepatocellular carcinoma progression. Biomed Res Int. 2018;2018:6784607. https://doi.org/10.1155/2018/6784607.
Liz J, Esteller M. lncRNAs and microRNAs with a role in cancer development. Biochim Biophys Acta. 2016;1859(1):169–76. https://doi.org/10.1016/j.bbagrm.2015.06.015.
Zhang Y, Liu X, Li Q. lncRNA LINC00460 promoted colorectal cancer cells metastasis via miR-939-5p sponging. Cancer Manag Res. 2019;11:1779–89. https://doi.org/10.2147/CMAR.S192452.
Du S, Hu W, Zhao Y, Zhou H, Wen W, Xu M, et al. Long non-coding RNA MAGI2-AS3 inhibits breast cancer cell migration and invasion via sponging microRNA-374a. Cancer Biomark. 2019;24(3):269–77. https://doi.org/10.3233/CBM-182216.
Wang G, Pan J, Zhang L, Wei Y, Wang C. Long non-coding RNA CRNDE sponges miR-384 to promote proliferation and metastasis of pancreatic cancer cells through upregulating IRS1. Cell Prolif. 2017. https://doi.org/10.1111/cpr.12389.
Sun H, He L, Ma L, Lu T, Wei J, Xie K, et al. LncRNA CRNDE promotes cell proliferation, invasion and migration by competitively binding miR-384 in papillary thyroid cancer. Oncotarget. 2017;8(66):110552–65. https://doi.org/10.18632/oncotarget.22819.
Bai PS, Xia N, Sun H, Kong Y. Pleiotrophin, a target of miR-384, promotes proliferation, metastasis and lipogenesis in HBV-related hepatocellular carcinoma. J Cell Mol Med. 2017;21(11):3023–43. https://doi.org/10.1111/jcmm.13213.
Wang YX, Chen YR, Liu SS, Ye YP, Jiao HL, Wang SY, et al. MiR-384 inhibits human colorectal cancer metastasis by targeting KRAS and CDC42. Oncotarget. 2016;7(51):84826–38. https://doi.org/10.18632/oncotarget.12704.
Eom S, Kim Y, Park D, Lee H, Lee YS, Choe J, et al. Histone deacetylase-3 mediates positive feedback relationship between anaphylaxis and tumor metastasis. J Biol Chem. 2014;289(17):12126–44. https://doi.org/10.1074/jbc.M113.521245.
Zheng J, Liu X, Wang P, Xue Y, Ma J, Qu C, et al. CRNDE promotes malignant progression of glioma by attenuating miR-384/PIWIL4/STAT3 Axis. Mol Ther. 2016;24(7):1199–215. https://doi.org/10.1038/mt.2016.71.
Nakamura H, Izumoto Y, Kambe H, Kuroda T, Mori T, Kawamura K, et al. Molecular cloning of complementary DNA for a novel human hepatoma-derived growth factor. Its homology with high mobility group-1 protein. J Biol Chem. 1994;269(40):25143–9.
Nakamura H, Kambe H, Egawa T, Kimura Y, Ito H, Hayashi E, et al. Partial purification and characterization of human hepatoma-derived growth factor. Clin Chim Acta. 1989;183(3):273–84.
Chen SC, Kung ML, Hu TH, Chen HY, Wu JC, Kuo HM, et al. Hepatoma-derived growth factor regulates breast cancer cell invasion by modulating epithelial–mesenchymal transition. J Pathol. 2012;228(2):158–69. https://doi.org/10.1002/path.3988.
Guo Z, He Y, Wang S, Zhang A, Zhao P, Gao C, et al. Various effects of hepatoma-derived growth factor on cell growth, migration and invasion of breast cancer and prostate cancer cells. Oncol Rep. 2011;26(2):511–7. https://doi.org/10.3892/or.2011.1295.
Acknowledgements
This work was supported by the Medicine and Health Science Technology Development Projects of Shandong Province (No. 2016WSO749).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Ethical consent was granted from the Medical Ethics Committee of Zibo Central Hospital for studies involving patients. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Ethical approval of the animal study was granted by the Animal Ethics Committee of Zibo Central Hospital.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1 si-SNHG3-1 inhibits breast cancer cell proliferation, migration and invasion
in vitro.(a) qRT-PCR verified the interference efficiency of SNHG3-1 in breast cancer cell lines. (b) A CCK-8 assay was performed to measure the proliferation of breast cancer cells after transfection with si-SNHG3-1 or si-NC. (c) A colony formation assay was performed to measure the proliferation of breast cancer cells after transfection. (d) Transwell assays were performed to determine the effects of SNHG3 knockdown on breast cancer cell migration and invasion. The experiments were performed independently in triplicate, and the data are presented as the mean ± SEM, ***P <0.001. (JPEG 858 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Ma, Q., Qi, X., Lin, X. et al. LncRNA SNHG3 promotes cell proliferation and invasion through the miR-384/hepatoma-derived growth factor axis in breast cancer. Human Cell 33, 232–242 (2020). https://doi.org/10.1007/s13577-019-00287-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-019-00287-9