
Abstract
Auxin response factors (ARFs) is one of common transcription factor (TF) family found in plants. By binding to its specific target in promoters, they regulate key genes related to auxin and mediating plant responses. So far, ARF family TFs have been identified in some plants, but no information is available regarding ARF genes in barley. In this study, we identified 20 ARF genes (HvARF) in the barley genome. Based on the comparative phylogenetic analysis, HvARFs were divided into six major Classes (I–VI). Duplication analysis revealed that four segmental and one tandem duplication contributed to the expansion of HvARF genes which were preferentially distributed at distal parts of the chromosomes. Mining of cis-acting regulatory elements in the promoter resulted that HvARF genes are mainly associated with hormone, stress and light responsiveness. In addition, they involve in miRNA-mediated gene regulation in which seven HvARF members were potentially targeted by five different miRNAs. Among those, hvu-miR160 simultaneously targets Class II ARF members (HvARF14, HvARF15, HvARF16, and HvARF17). Besides, the expression pattern of 20 HvARFs was assessed in different embryonic development and stress-induced (heat, drought, salinity, and excess boron) conditions, which pointed out that HvARFs functionally involve in developmental stages and stress adaptation of barley, by regulating the auxin-mediated genes. The results represented here provide comprehensive infomation on ARF family genes and could provide a framework for further cloning and functional verification of HvARF members for the purposes of crop improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Auxin, the first plant hormone to be discovered, has many roles in plant growth and development. It mediates elongation of stem and root growth, enlargement of fruits and tubers, promotion of cell division. It regulates expression of some classes of genes such as Aux/IAA, Gretchen Hagen3 (GH3), and SMALL AUXIN UP RNA (SAUR) (Abel and Theologis 1996). In addition, auxin response factor (ARF) transcription factors (TFs) are important players in the regulation of auxin-mediated biological processes by binding auxin response elements (AuxREs) locate in the promoter region of auxin-responsive genes. Specifically, ARF TFs bind to TGTCTC motif or its variations such as TGTCCC, TGTCAC, and TGTCGG, and cause to repression or activation of these genes (Ulmasov et al. 1997; Guilfoyle and Hagen 2007; Boer et al. 2014).
As a protein, ARFs interact with DNA as well as proteins by forming a homodimer or heterodimer structure. Protein–protein and protein-DNA interaction of ARF protein is maintained by its three characteristic region: (1) an N-terminal B3-type DNA binding-domain (DBD) which interact with AuxREs found in the upstream promoter region of auxin responsive genes; (2) a C-terminal dimerization domain (CTD or domain III/IV) which is essential for ARF-Aux/IAA and/or ARF–ARF dimerization (Kim et al. 1997; Shen et al. 2010); and (3) a variable middle region (MR) that is functional in activation or repression of auxin-responsive genes (Ulmasov et al. 1997; Tiwari et al. 2003; Leyser 2018). B3-type DBD is classified as a plant-specific domain that is found in a variety of plant transcription factors, including ARFs. The domain is indispensable for ARF-AuxREs interaction. Moreover, ARF proteins can bind DNA as a dimer, monomer or heterodimer (4) which can change promoter activity accordingly. Depending on the amino acid composition of MR, ARF proteins act as activator or repressor i.e., glutamine, serine and leucine-rich MR could cause activation; serine, proline, leucine and glycine-rich MR could lead to repression. Also, ARF expression can be regulated by auxin status of the cell. Under low auxin conditions, Aux/IAA interacts with ARF and the formed complex represses the auxin-responsive gene expression. In opposite, high auxin level causes Aux/IAA release from ARFs and forms a complex with TIR1 and SCF proteins which direct it to 26S proteasomal degradation. Then, ARF proteins alone activates expression of auxin-responsive genes [reviewed by Leyser (2018) and Korasick et al. (2015)].
Sequencing technologies let comprehensive genome and transcriptome-wide analysis of multigene families (Ablazov and Tombuloglu 2016; Tombuloglu et al. 2013; 2015; 2016). In the last decade, ARF TFs were intensively identified in a variety of plant species i.e., Arabidopsis, rice, Populus, tomato, grape etc. Recently, a comprehensive evolutionary analysis of ARF proteins from angiosperm genomes was carried out (Finet et al. 2012). However, there is any report regarding ARF gene family of barley, a widely cultivated crop species in the world. Its first draft genome sequence was released in 2012 (IBSC 2012) and finally full genome was mapped in 2016 (Mascher et al. 2017). In this article, we aimed to find and characterize ARF family members in barley with their comprehensive characteristics i.e. conserved domain/residue, cis-acting regulatory elements, potential miRNA targets, and tissue/organ-specific expressions by using bioinformatics tools.
Matherials and methods
Mining of ARF genes in the barley genome
Barley (Hordeum vulgare L.) nucleotide, CDS (coding DNA sequence) and protein sequences were retrieved from barley genome database, IBSC (International barley sequence consortium) (Morex 50× WGS sequence) (ftp://ftpmips.helmholtz-muenchen.de/plants/barley/public_data/) (IBSC 2012). Barley ARF sequences were searched using a local BlastP program (version 2.2.30) (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (Altschul et al. 1997) against 23 Arabidopsis ARF sequences as a query (Wang et al. 2007). Redundant and low-confident sequences were removed from the total ARF list. The sequences were further analyzed using Pfam (release 31.0) (http://pfam.xfam.org/) (Finn et al. 2015) and InterproScan (release 5.14-53.0) (http://www.ebi.ac.uk/interpro/) (Jones et al. 2014) domain finders. Domain hits lower than e-10 was considered significant. Barley HORVU Ids, from the barley full genome sequence (Mascher et al. 2017), were obtained from the BlastP server of IPKBarley (http://webblast.ipk-gatersleben.de/barley_ibsc), by searching the barley genome accession numbers as a query against to `Barley HC proteins 2016` dataset. Protein identity score and percentages with an e-value score were noted to compare draft (IBSC 2012) and full genome (Mascher et al. 2017) releases.
Chromosome, duplication and sequence analysis of barley ARFs
The information of barley ARFs was obtained from several databases. Representative gene IDs were received from “barley_ALLgenes_MIPS_15Jul14_MAPPINGinfo” file (IBSC 2012); contig numbers were obtained from POPSEQ data generated by IPK (Comadran et al. 2012; Mascher et al. 2013; Ariyadasa et al. 2014); chromosome location and intron–exon structure was downloaded from EnsemblPlants database (http://plants.ensembl.org/Hordeum_vulgare/Info/Annotation); physiochemical characteristics of ARF proteins were determined in ProtParam server (http://web.expasy.org/protparam/) (Gasteiger et al. 2005). GSDS (Gene Structure Display Server) was used to compute intron and exon boundaries (http://gsds.cbi.pku.edu.cn/) (Hu et al. 2014). To identify gene duplication event that possibly cause gene expansion, HvARF members were aligned by ClustalOmega program and then the following parameters were adopted to detect duplicated gene pairs: the alignment of the coding nucleotide sequences covered 70% of the longest genes and the amino acid identity between the sequences was > 70% (Yang et al. 2008; Bostancioglu et al. 2018).
Putative miRNA target and cis-acting element analyses
We retrieved barley mature miRNA sequences from databases and published articles: 71 sequences from miRbase v.20 (http://www.mirbase.org/) (Kozomara and Griffiths-Jones 2014), 23 sequences from the Plant microRNA database (http://bioinformatics.cau.edu.cn/PMRD/) (Zhang et al. 2010), and 40 sequences from (Ozhuner et al. 2013) and (Ferdous et al. 2017). CDS with 5′ and 3′ UTR sequences of all HvARF genes were obtained from EnsemblPlant database and searched to extract putative miRNA targets by using psRNATarget server with 3.5 maximum expectation score, others adjusted to default parameters (http://plantgrn.noble.org/psRNATarget/?function=3) (Dai and Zhao 2011). Cis-acting elements were searched from the promoter regions of ARF genes by using online PlantCARE software (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (Lescot et al. 2002). 1000 bp of the 5′ flanking genomic DNA (upstream) sequence of each ARF genes was retrieved from EnsemblPlants database (http://plants.ensembl.org/Hordeum_vulgare/Info/Index) and used for the analysis.
Multiple sequence alignment and phylogenetic analysis of ARFs
The protein sequences were aligned by the ClustalOmega tool (http://www.ebi.ac.uk/Tools/msa/clustalomega/) and conserved domains and critical residues for protein–protein interaction were further examined. The new nomenclature for barley ARF proteins/genes were given according to ClustalOmega alignment order. Aligned protein sequences were further used for phylogenetic analysis. In addition to 20 barley ARF protein sequences, Arabidopsis (23), Brassica rapa (31), Zea mays (36), Solanum lycopersicum (21) and Vitis vinifera (19) ARF proteins were collected and a comprehensive and comparative phylogenetic analysis was conducted. Phylogenetic trees were produced using MEGA 6 (http://www.megasoftware.net) (Tamura et al. 2013) according to neighbor-joining (NJ) and maximum likelihood (ML) methods based on Poisson correction model. The parameters were adjusted to Data subset to use, complete deletion; replication, bootstrap analysis with 1000 replicates. Finally, the cut-off value for the condensed tree was set to 50%. The further tree analysis was carried out by using FigTree program (version 1.4.2) (http://tree.bio.ed.ac.uk/software/Figtree/).
Digital expression analysis of barley ARFs
Spatial and temporal expression patterns of 20 HvARF genes from different development stages and organs (root, shoot, tillers, grain, embryos, caryopsis (5 and 15 mm long), inflorescence (young and developing), and internode) were represented in a heat map. The expression values (FPKM: Fragments Per Kilobase of transcript per Million mapped reads) were retrieved from BARLEX (Colmsee et al. 2015) and PGSB (Plant genome and System biology) (http://pgsb.helmholtz-muenchen.de/plant/index.jsp) databases. Besides, barley GEO datasets, accession number GSE82134 and GSE66024, were used to monitor the expression profiles of HvARFs under heat stress (Pacak et al. 2016) and seed germination stages into 24 and 48 h periods (Zhang et al. 2016), respectively. Expression patterns were visualized by using PermutMatrix v1.9.3 program (http://www.atgc-montpellier.fr/) (Caraux and Pinloche 2005). Dataset was adjusted to interaction term (row/column), and all the other parameters were fixed as default. Also, Genevestigator platform was used to evaluate the HvARF gene expressions under different type of stress conditions such as; drought, salinity, heat, and excess boron (https://genevestigator.com/gv/) (Zimmermann et al. 2004). Hierarchical clustering mode with Euclidian distance parameter was set to monitor the expression levels with log2 ratio calculation based on a control group.
Results and discussion
ARF genes were identified in the barley genome
To identify barley ARFs, a local BlastP search was carried out using 23 AtARF proteins as a query. High confidence (HC) protein sequence information was used as a subject. The hits higher than e-10 were rejected to determine candidate HvARFs. Furthermore, characteristic domains for ARF proteins were mined from those proteins to validate barley ARFs. The proteins containing at least B3 (PF02362) and Auxin_resp (PF06507) domains were selected and identified as candidate ARF member in barley. Other than those domains, 11 HvARF sequences harbored an additional conserved domain, which is AUX/IAA (PF02309) (Fig. 1c; Table S1). To determine the significance of each domain <e-10 criteria was used. The conserved domains were also checked with InterProScan analysis. At the end, 20 canonical, non-redundant HvARF members were identified and characterized in barley. The nomenclature of new ARF proteins was set according to their alignment order in the ClustalOmega analysis.

Neighbor-joining (NJ) tree of 20 HvARF proteins (a), their coding gene (b), and protein domain structures (c). B3 and Auxin_resp domains are common in all proteins. All Class I and II members exclude Aux/IAA domain. The gene and protein size scales were positioned below the images. The phylogenetic tree based on maximum likelihood (ML) analysis was given in Fig. S2
Also, we checked and validated these protein sequences from the latest genome data (Barley HC Proteins May 2016, Mascher et al. 2017) and observed that some ARF members were excluded from the protein dataset released in 2016. For instance, ARF16 (MLOC_64795.1) involve in the proteome release of 2012 (HC_genes_AA_Seq_2012; (14)). However, it was represented by the protein (HORVU7Hr1G101270.6) with percentage of % 62 in 2016 HC proteome dataset, showing inconsistency between the protein data of 2012 and 2016 releases. Also, it was noted that HORVU7Hr1G101270.6 and MLOC_64795.1 were different proteins based on their chromosome location and protein characteristics. In our analyses, we used the early genome release data (MLOC_64795.1) to take ARF16 into account as ARF member. The nucleotide sizes of each barley chromosome were retrieved from EnsemblPlants database and chromosome location of each ARF member were shown according to the latest genetic map which is based on chromosome conformation capture sequencing (Hi-C) strategy (Fig. 2). ARF16* shows a new protein (HORVU7Hr1G101270.6) based on the latest barley genome release (2016), which has the highest identity score (62%) to HvARF16 (MLOC_64795.1) from barley draft genome release in 2012. Also, remaining ARF members were checked in both databases, and all of them matched to the corresponding proteins (% 99 or 100) with an expected value of 0 (Table 1). HvARF protein sequences were listed in Table S4.

Distribution of 20 HvARF genes along the chromosomes with their tandem and segmental duplications. Segmentally duplicated gene pairs are shown with a dashed line and labeled with the same background color. Dark and light blue chromosomes pairs show susceptible regions prone to segmental duplication. Green and red arrows represent the gene directions either negative or positive strand, respectively. Chromosome sizes were represented with megabase pairs (Mbp). The number of HvARF genes located on each chromosome is stated below the chromosome number in parenthesis (color figure online)
Barley genes have been represented with several gene IDs by different genome datasets. In our study, we listed the HvARF TFs with their all representative gene IDs based on IBSC (2012), POPSEQ (Comadran et al. 2012; Mascher et al. 2013; Ariyadasa et al. 2014), and the latest genome information (Mascher et al. 2017) (Table 1). Also, UniProt proteome entries of each HvARF members were represented. HvARF ORF lengths ranged from 3528 (HvARF02) to 1164 bp (HvARF15), and molecular weights ranged from 131.22 (HvARF02) to 50.34 kDa (HvARF05), and pI values ranged from 8.92 (HvARF14) to 5.77 (HvARF07) (Table 1). Based on exon and intron structures, exon numbers were well separated between the ARF Classes (I to VI), for instance, average exon number of HvARF genes in Class I and V were 13 (Fig. 1b; Table 1). However, Class II members had 2 or 3 exons, showing the structural divergence of ARF genes.
Chromosomal distribution and gene duplication analysis of HvARF genes
Chromosome distribution of HvARF genes was shown in Fig. 2 where they dispersed in all barley chromosomes (Chr) except chr4H. Chromosome 3H and 7H harbored four ARF members and chr2H included five, which is the largest number of ARF genes represented in a single chromosome. To find out putative gene duplications among HvARF gene pairs, we conducted a duplication analysis based on protein similarity and gene distribution along the chromosomes. Accordingly, the gene pairs with > 70% amino acid and nucleotide identity score were selected as potential duplicated genes (Table S5). Then, these gene pairs were checked for their chromosomal positions. The results revealed that four segmental and one tandem duplication contributed to the expansion of ARF family in barley. Accordingly, segmental duplications appeared between ARF05/ARF06, ARF05/ARF07, ARF05/ARF08, and ARF01/ARF02 genes. It is interesting that ARF05 is the crosspoint for the segmental duplication of three genes (ARF06, ARF07, and ARF08) by rooting the gene expansion from chr6H or vice versa. Also, these genes were included in the same phylogenetic group (Class V or AtARF6/8-like) (Figs. 1, 3; Fig. S2) and well-separated from the other ARF members, shows that segmental duplication specifically contributed to the expansion of Class V in barley. Moreover, duplication event between ARF01/ARF02 and ARF05/ARF06 genes pointed out that a large segment of chromosome duplication had been appeared between chr6H and chr7H, where the direction of the genes (+ or -) agree for a reverse segmental duplication. Additionally, tandem duplication in chr3H resulted to appear ARF10 or ARF11 genes (Fig. 2). Expansion of ARF family members by gene duplication may cause to gain new functions (neofunctionalization) which can alter the genetic control of ARF-mediating cellular mechanism. According to a recent scenario based on the full genome sequence of barley proposed by Keller and Krattinger (2017), barley genes are well organized in the chromosomes where distal parts are enriched in rapidly evolving genes and prone to recombination than the proximal and interstitial regions. However, proximal part enriched in housekeeping genes has the low recombination frequency. In our study, ARF genes were found to be distributed generally in distal regions of the chromosomes. Also, duplications of ARF genes have occurred in those regions which is supporting the chromosomal compartmentalization scenario in the barley genome.

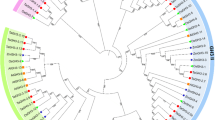
Comparative phylogenetic analysis of 20 HvARFs with Arabidopsis (23), Brassica rapa (31), Zea mays (36), Solanum lycopersicum (21) and Vitis vinifera (19) ARF sequences. Arabidopsis and barley ARFs was represented with red and blue colors, respectively. Six major ARF classes (Class I–VI) with their corresponding names for AtARFs were shown (color figure online)
Critical residues for DNA-ARF, ARF–ARF and Aux/IAA-ARF interactions
DNA interaction residues of ARF proteins were determined according to a previous report described by Boer et al. (2014). 20 HvARF members were aligned and identical, conserved, semi-conserved and non-conserved residues for DNA contact were shown (Fig. S3). Ser (S), His (H), Gly (G), Ser (S), Arg (R) and Arg (R), Pro (P), Arg (R), Thr (T), Ser (S) were the first and the second DNA contacting loop residues, respectively. His (H) and Thr (T) were determined as semi-conserved residues, in which Gly (G) and Ser (S) were found to be substituted in some of ARF members, respectively (Fig. S3). These were the critical residues of the DBD domain for DNA contact.
In the C-terminus of ARF proteins, a typical CTD (type-I/II Bem1p (PB1) and Phox domains) were harbored that provides positive and negative electrostatic surfaces for protein–protein interaction (Korasick et al. 2014). To uncover the protein–protein interacting residues of ARFs, i.e. ARF-Aux/IAA or ARF–ARF, Motif III and IV (CTD) were further evaluated. Conserved Lys (K) residue (type II, gain a basic surface) resided in motif III of the selected HvARF members. Also, an OPCA-like motif [type I, expose an acidic surface and makes salt bridges with basic residues, especially with Lys (K) and resulting homo- or hetero-dimerization of ARFs (Sumimoto et al. 2007)], was found in motif IV (Fig. S4). OPCA-like and conserved Lys (K) could enable ARF–ARF or ARF-Aux/IAA dimerization and could change the auxin-responsive gene expression during auxin signaling. However, out of 20 HvARF proteins, only 13 members harbor Motif III and IV domains (Fig. S1 and S4), pointed out that those members may not be involved in dimerization. Besides, motif III and IV region composed of five β-sheet (β1–β5) and three α-helix (α1–α3). The linker loop between the two motifs exhibited sequence and length variance (Fig. S3), which may alter ARF and protein binding selectivity. To detect Aux/IAA interacting ARFs, further studies could be focused to identify barley Aux/IAA family sequences.
Phylogenetic analysis of barley ARFs
HvARF proteins were aligned and subdivided into six classes based on topology and sequence similarity analysis by using NJ (Neighbor-joining) and maximum likelihood (ML) trees with 50% bootstrap support (Fig. 1a). In addition, an ML (maximum likelihood) tree was constructed and it was observed that the topologies of both trees were identical to each other (Fig. S2), indicating the two methods were in good agreement. Combining the HvARF protein sequences with Arabidopsis, B. rapa, Z. mays, S. lycopersicum, and V. vinifera ARFs, a comparative phylogenetic analysis was carried out (Fig. 3). Like the barley phylogeny results, the constructed tree separated them into six major classes (I–VI). Likewise, ARF sequences from distinct plants have been categorized to the same Classes (Table 2). Each HvARF members were found to be dispersed to their exact groups located in the barley phylogeny analysis result, indicating the comprehensive phylogenetic analysis agrees with that of barley results. ARF family TFs have been extensively studied in Arabidopsis in which ARF5 and ARF1 were found to exhibit antagonistic function (Rademacher et al. 2011). In the phylogenetic classification, AtARF5 and HvARF09 were resided in the same class (Class IV), and AtARF1 and HvARF12 were grouped in the same clade of Class III, suggesting possible antagonistic functionalization of HvARF09 and HvARF12 in barley. However, a functional analysis is essential to clarify their exact roles.
Cis-acting regulatory elements and putative miRNA target analyses
Cis-regulatory elements (CREs) are critical sequences which are specifically targeted by transcription factors to regulate gene expression. So far, many CREs have been determined and their potential roles were identified. In this study, to understand the potential roles of HvARFs, we analyzed the promoter region of each HvARF genes by using PlantCARE software. Except for the two genes (HvARF09 and HvARF12), we listed all CREs found on the promoter site (Table S6). They were categorized into six major functional groups; hormone response, stress, circadian control, light responsiveness, promoter/enhancer element, and development/tissue specificity related elements (Fig. S5). Analyses revealed that promoter/enhancer elements (CAAT-box and TATA-box) were abundantly placed at the promoters of HvARFs in general. Also, light responsive and hormone response elements were the other most abundant functional categories. Hormone response elements were comprised of AuxRR-core and TGA-element which are directly associated with auxin-responsiveness. These results suggested that HvARF genes were mainly associated with hormone, stress, light responsiveness, as well as development and tissue-specific expressions. Also, some of the ARF genes could be related to the regulation of Circadian rhythm.
miRNAs regulate gene expression by post-translational modifications via mRNA cleavage or translational inhibition. To detect potential regulation of HvARFs by miRNA, we analyzed cDNA sequences including 5′ and 3′ untranslated regions (UTRs), and the coding regions of all ARF members in barley. We used the psRNATarget program to detect miRNA targets. Maximum expectation score was settled to 3.5 (instead of 5) to maximize the probability to capture the highest miRNA-target complementary. The analyses resulted that five different miRNAs target seven HvARF mRNAs at 11 prediction sites (Fig. 4; Table S3). Detected miRNAs bind to coding or non-coding regions (UTR) of ARF transcripts that triggers cleavage or translation inhibition. For instance, HvARF19 was targeted by hvu-miR5049c and hvu-miR6197, which promote cleavage and translational inhibition, respectively. Also, HvARF19 was targeted by three different miRNAs (hvu-miR1436, hvu-miR5049c, and hvu-miR6197). hvu-miR5049c has potential to inhibit three ARF members (HvARF01, HvARF03, and HvARF19) by cleavage. Notably, miR6197 targets HvARF19 mRNA from two complementary sequences located in the same exon (exon 1). Interestingly, hvu-miR160 simultaneously targets four ARF members (HvARF14, HvARF15, HvARF16, and HvARF17) which are the members of Class II (AtARF10/16/17-like) subgroup. It seems that these ARFs could functionally be regulated by miR160 and may have a cooperative role in the cell. Previous reports support this idea, for instance miR160 targets ARF10, ARF16, and ARF17 (Class II ARFs) in Arabidopsis and directs posttranscriptional regulation during growth and development processes (Mallory et al. 2005), i.e. miR160 targeted ARF10 and ARF16 in Arabidopsis controls root cap cell formation (Wang et al. 2005), and in rice miR160 negatively regulates of its target (OsARF18) which caused alteration in auxin signaling and development (Huang et al. 2016).

miRNAs are potentially targeting HvARFs. Putative miRNA-ARF complementary may repress the expression of some HvARFs via either directing cleavage or translation inhibition. Duplicated HvARFs or hvu-miRNAs were labeled with the same color code (color figure online)
Expressions of HvARFs in tissues and stress conditions
ARF family transcript accumulation was assessed in 16 different developmental stages of barley which gives deep information to understand possible organ or tissue-specific functionalities of HvARFs separately. RNA-Seq-based expressions were represented with three or two replicates retrieved from BARLEX and PGSB databases. Heatmap analysis revealed that Class I, II, and V members clustered together, which shows similar expression patterns among the selected tissues. Also, ARFs repressed or activated in specific tissues showed their spatial and temporal expression (Fig. S6). In addition, accumulation of ARF transcripts during seed germination stages (24 h and 48 h) from four barley cultivars (Bass, Harrington, Stirling, and Baudin) were assessed (from GEO dataset, GSE66024). The results showed that in general ARF expressions were dramatically changed during germination periods (Fig. S9a). This implies the importance of HvARF TFs for the development of embryo and organs. Log2 calculated expression data represented in Table S2.
Together with, expression level of HvARF genes were assessed under varied stress conditions such as drought with three different water content stages (7, 19, and 38% SWC), heat (0.5, 3 and 6 h), salinity (root and shoot), and excess boron (5 and 10 mM boric acid). The data obtained for 10 HvARF members and represented in Fig. S8. The results pointed out that some ARF members were highly expressed under drought stress condition, for instance, HvARF10 and HvARF11. In addition, HvARF15 and HvARF16 were the heat stress responsive transcripts where their expression was downregulated. However, expression of these genes was tissue-specific and reversely altered at the root and shoot tissues after 1 h of heat treatment (GEO dataset: GSE82134) (Fig. S7b). Also, we found that salt and boron stress did not significantly alter ARF expressions.
We identified 20 HvARF genes in the latest genome release of barley. Chromosome distribution of HvARFs showed that they were preferentially located in the distal parts of the chromosomes where duplication frequency of the genes was higher than the proximal and interstitial parts. In those regions, segmental and tandem duplications took place and contributed to the expansion of HvARF family. Critical residues of HvARFs for protein–protein interaction and DNA binding were determined which are critical for ARF functionality. Analysis of cis-acting elements revealed that hormone response, stress, circadian control, light responsiveness, promoter/enhancer element, and development/tissue specificity related elements were abundantly placed in the promoter region of HvARF genes. In addition, post-transcriptional modification of HvARFs via miRNAs were assessed and we found that seven HvARFs were potentially targeted by five different miRNAs that can regulate auxin-mediated biological processes. Interestingly, hvu-miR160 simultaneously targets Class II members (HvARF14, HvARF15, HvARF16, and HvARF17), whose expression pattern were similar to each other in 16 tissues represented in this study. In addition, RNA-Seq-based expression patterns of HvARFs were determined in various stress conditions, revealed that ARF family members involve in abiotic stress tolerance and adaptation. Those results revealed a comprehensive framework to understand the biological roles of ARFs in both barley and plant species. Also, it helps to uncover auxin-mediated gene regulatory mechanisms in plants. Moreover, the data represented here can be useful for further functional characterization studies of ARFs based on overexpression, RNAi or genome editing methods such as CRISPR-Cas9.
Abbreviations
- ARF:
-
Auxin response factors
- AuxREs:
-
Auxin response elements
- IBSC:
-
The International Barley Genome Sequencing Consortium
- CDS:
-
Coding DNA sequence
- CREs:
-
Cis-regulatory elements
- miRNA:
-
MicroRNA
- TF:
-
Transcription factor
References
Abel S, Theologis A (1996) Early genes and auxin action. Plant Physiol 111(1):9
Ablazov A, Tombuloglu H (2016) Genome-wide identification of the mildew resistance locus O (MLO) gene family in novel cereal model species Brachypodium distachyon. Eur J Plant Pathol 145(2):239–253
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ariyadasa R, Mascher M, Nussbaumer T, Schulte D, Frenkel Z, Poursarebani N et al (2014) A sequence-ready physical map of barley anchored genetically by two million single-nucleotide polymorphisms. Plant Physiol 164(1):412–423
Boer DR, Freire-Rios A, van den Berg WA, Saaki T, Manfield IW, Kepinski S et al (2014) Structural basis for DNA binding specificity by the auxin-dependent ARF transcription factors. Cell 156(3):577–589
Bostancioglu SM, Tombuloglu G, Tombuloglu H (2018) Genome-wide identification of barley MCs (metacaspases) and their possible roles in boron-induced programmed cell death. Mol Biol Rep. https://doi.org/10.1007/s1103
Caraux G, Pinloche S (2005) PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21(7):1280–1281
Colmsee C, Beier S, Himmelbach A, Schmutzer T, Stein N, Scholz U, Mascher M (2015) BARLEX—the barley draft genome explorer. Mol Plant 8:964–966
Comadran J, Kilian B, Russell J, Ramsay L, Stein N, Ganal M et al (2012) Natural variation in a homolog of Antirrhinum CENTRORADIALIS contributed to spring growth habit and environmental adaptation in cultivated barley. Nat Genet 44(12):1388–1392
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39(Web Server issue):W155–W159
Ferdous J, Sanchez-Ferrero JC, Langridge P, Milne L, Chowdhury J, Brien C, Tricker PJ (2017) Differential expression of microRNAs and potential targets under drought stress in barley. Plant Cell Environ 40(1):11–24
Finet C, Berne-Dedieu A, Scutt CP, Marlétaz F (2012) Evolution of the ARF gene family in land plants: old domains, new tricks. Mol Biol Evol 30(1):45–56
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL et al (2015) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44(D1):D279–D285
Gasteiger E, Hoogland C, Gattiker A et al (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, Clifton, pp 571–607
Guilfoyle TJ, Hagen G (2007) Auxin response factors. Curr Opin Plant Biol 10(5):453–460
Hagen G, Guilfoyle T (2002) Auxin-responsive gene expression: genes, promoters and regulatory factors. Plant Mol Biol 49(3–4):373–385
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G (2014) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31(8):1296–1297
Huang J, Li Z, Zhao D (2016) Deregulation of the OsmiR160 target gene OsARF18 causes growth and developmental defects with an alteration of auxin signaling in rice. Sci Rep 6:29938
Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C et al (2014) InterProScan5: genome-scale protein function classification. Bioinformatics 30(9):1236–1240
Keller B, Krattinger SG (2017) Plant science: genomic compartments in barley. Nature 544(7651):424–425
Kim J, Harter K, Theologis A (1997) Protein–protein interactions among the Aux/IAA proteins. Proc Natl Acad Sci USA 94(22):11786–11791
Korasick DA, Westfall CS, Lee SG, Nanao MH, Dumas R, Hagen G et al (2014) Molecular basis for AUXIN RESPONSE FACTOR protein interaction and the control of auxin response repression. Proc Natl Acad Sci USA 111(14):5427–5432
Korasick DA, Jez JM, Strader LC (2015) Refining the nuclear auxin response pathway through structural biology. Curr Opin Plant Biol 27:22–28
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42(Database issue):D68–D73
Lescot M, Déhais P, Thijs G, Marchal K et al (2002) PlantCARE a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucl Acids Res 30(1):325–327
Leyser O (2018) Auxin signaling. Plant Physiol 176(1):465–479
Mallory AC, Bartel DP, Bartel B (2005) microRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 17(5):1360–1375
Mascher M, Muehlbauer GJ, Rokhsar DS, Chapman J, Schmutz J, Barry K et al (2013) Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). Plant J 76(4):718–727
Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544(7651):427–433
Mun JH, Yu HJ, Shin JY, Oh M, Hwang HJ, Chung H (2012) Auxin response factor gene family in Brassica rapa: genomic organization, divergence, expression, and evolution. Mol Genet Genom 287(10):765–784
Ozhuner E, Eldem V, Ipek A, Okay S, Sakcali S, Zhang B et al (2013) Boron stress-responsive microRNAs and their targets in barley. PLoS ONE 8(3):e59543
Pacak A, Barciszewska-Pacak M, Swida-Barteczka A, Kruszka K et al (2016) Heat stress affects Pi-related genes expression and inorganic phosphate deposition/accumulation in barley. Front Plant Sci Q 7:926
Rademacher EH, Möller B, Lokerse AS, Llavata-Peris CI, van den Berg W, Weijers D (2011) A cellular expression map of the Arabidopsis AUXIN RESPONSE FACTOR gene family. Plant J 68(4):597–606
Shen C, Wang S, Bai Y, Wu Y, Zhang S, Chen M et al (2010) Functional analysis of the structural domain of ARF proteins in rice (Oryza sativa L.). J Exp Bot 61(14):3971–3981
Shen C, Yue R, Sun T, Zhang L, Xu L, Tie S, Wang H, Yang Y (2015) Genome-wide identification and expression analysis of auxin response factor gene family in Medicago truncatula. Front Plant Sci 6:73
Sumimoto H, Kamakura S, Ito T (2007) Structure and function of the PB1 domain, a protein interaction module conserved in animals, fungi, amoebas, and plants. Sci STKE 2007(401):re6–re6
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
The International Barley Genome Sequencing Consortium (IBSC) (2012) A physical genetical and functional sequence assembly of the barley genome. Nature 491:711–717
Tiwari SB, Hagen G, Guilfoyle T (2003) The roles of auxin response factor domains in auxin-responsive transcription. Plant Cell 15(2):533–543
Tombuloglu H, Kekec G, Sakcali MS, Unver T (2013) Transcriptome-wide identification of R2R3-MYB transcription factors in barley with their boron responsive expression analysis. Mol Genet Genom 288(3–4):141–155
Tombuloglu G, Tombuloglu H, Sakcali MS, Unver T (2015) High-throughput transcriptome analysis of barley (Hordeum vulgare) exposed to excessive boron. Gene 557(1):71–81
Tombuloglu H, Ozcan I, Tombuloglu G, Sakcali S, Unver T (2016) Aquaporins in boron-tolerant barley: identification characterization and expression analysis. Plant Mol Biol Rep 34(2):374–386
Ulmasov T, Hagen G, Guilfoyle TJ (1997) ARF1, a transcription factor that binds to auxin response elements. Science 276(5320):1865–1868
Van Ha C, Le DT, Nishiyama R, Watanabe Y, Sulieman S, Tran UT et al (2013) The auxin response factor transcription factor family in soybean: genome-wide identification and expression analyses during development and water stress. DNA Res 20(5):511–524
Wan S, Li W, Zhu Y, Liu Z, Huang W, Zhan J (2014) Genome-wide identification, characterization, and expression analysis of the auxin response factor gene family in Vitis vinifera. Plant Cell Rep 33(8):1365–1375
Wang JW, Wang LJ, Mao YB, Cai WJ, Xue HW, Chen XY (2005) Control of root cap formation by microRNA-targeted auxin response factors in Arabidopsis. Plant Cell 17(8):2204–2216
Wang D, Pei K, Fu Y, Sun Z, Li S, Liu H et al (2007) Genome-wide analysis of the auxin response factors (ARF) gene family in rice (Oryza sativa). Gene 394(1):13–24
Wang Y, Deng D, Shi Y, Miao N, Bian Y, Yin Z (2012) Diversification, phylogeny and evolution of auxin response factor (ARF) family: insights gained from analyzing maize ARF genes. Mol Biol Rep 39(3):2401–2415
Wu J, Wang F, Cheng L, Kong F, Peng Z, Liu S et al (2011) Identification, isolation and expression analysis of auxin response factor (ARF) genes in Solanum lycopersicum. Plant Cell Rep 30(11):2059
Yang S, Zhang X, Yue JX, Tian D, Chen JQ (2008) Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol Genet Genom 280(3):187–198
Zhang Z, Yu J, Li D, Zhang Z, Liu F, Zhou X, Wang T, Ling Y, Su Z (2010) PMRD: plant microRNA database. Nucl Acids Res 38(Database issue):D806–D813
Zhang Q, Zhang X, Wang S, Tan C et al (2016) Involvement of alternative splicing in barley seed germination. PLoS ONE 11(3):e0152824
Zimmermann P, Hirsch-Hoffmann M, Hennig L, Gruissem W (2004) GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol 136(1):2621–2632
Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Human and animals rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tombuloglu, H. Genome-wide analysis of the auxin response factors (ARF) gene family in barley (Hordeum vulgare L.). J. Plant Biochem. Biotechnol. 28, 14–24 (2019). https://doi.org/10.1007/s13562-018-0458-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-018-0458-6