
Abstract
Background
PRMT5 is a type II protein arginine methyltransferase that methylates histone or non-histone proteins. Arginine methylation by PRMT5 has been implicated in gene transcription, ribosome biogenesis, RNA transport, pre-mRNA splicing and signal transduction. High expression of PRMT5 has been observed in various cancers and PRMT5 overexpression has been reported to improve cancer cell survival, proliferation, migration and metabolism and to inhibit cancer cell apoptosis. In addition, PRMT5 has been found to be required for cancer stem cell survival, self-renewal and differentiation. Several microRNAs have been shown to regulate PRMT5 expression. As PRMT5 has oncogene-like properties, several PRMT5 inhibitors have been used to explore their efficacy as potential drugs for different types of cancer, and three of them are now being tested in clinical trials.
Conclusions
In this review, we summarize current knowledge on the role of PRMT5 in cancer development and progression, including its functions and underlying mechanisms. In addition, we highlight the rapid development of PRMT5 inhibitors and summarize ongoing clinical trials for cancer therapy. By affecting both tumor cells and the tumor microenvironment, PRMT5 inhibitors may serve as effective anti-cancer agents, especially when combined with immune therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
A common mechanism for cancer occurrence and development is epigenetic dysregulation. Epigenetic regulation involves the modification of DNA or histones via methylation, acetylation, phosphorylation, ubiquitination and alterations in chromatin remodeling, thereby affecting gene expression and function while retaining the intact DNA sequence [1]. Among these, arginine methylation is a key post-translational modification, first reported in 1971, which adds methyl groups to the nitrogen atoms of arginine residues in the polypeptide [2, 3]. The major regulators of arginine methylation include the nine members of the PRMT family, which have recently been found to be highly expressed in various solid and hematological tumors. Elevated PRMT expression correlates with a poor prognosis, although PRMT mutations are rare in tumors [4].
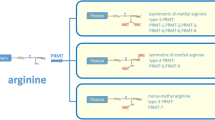
In eukaryotes, methylated arginines can be classified into three main forms, monomethylarginines (MMAs), asymmetric dimethylarginines (ADMAs) and symmetric dimethylarginines (SDMAs). In the PRMT family, type I PRMTs (PRMT1, 2, 3, 4, 6 and 8) catalyze MMA and ADMA synthesis, type II PRMTs (PRMT5 and PRMT9) catalyze MMA and SDMA synthesis, and type III PRMT (PRMT7) is capable of monomethylation. In fungi, there is another type IV PRMT that generates δ-NG-monomethylarginine [5]. Recently, with the development of structural biology and an increasing understanding of cancer biology, novel inhibitors of PRMT1 and PRMT5 have been developed, some of which are now entering phase I clinical trials [4]. In this review, we provide a summary of the structure, function and regulation of PRMT5 in cancer and describe how PRMT5 can be employed as a potential target for cancer therapy.
2 PRMT5 is a type II protein arginine methyltransferase
PRMT5 is the primary type II protein arginine methyltransferase that catalyzes MMA and SDMA synthesis. It is also referred to as histone synthetic lethal 7 (HSL7), Janus binding protein 1 (JBP1), Schizosaccharomyces pombe Shk1 kinase-binding protein 1 (SKB1), Capsuleen, or Drosophila arginine methyltransferase 5 (DART5). The PRMT5 gene was cloned from Saccharomyces cerevisiae and human in 1996–1999, and its catalytic domain was found to be highly conserved, though multiple PRMT5 splice variants can be found in human cells [6, 7]. PRMT5 is a two-domain protein. The N-terminal domain adopts a triosephosphate isomerase barrel structure and binds methylosome protein 50 (MEP50) as a requirement for its full methyltransferase activity, whereas the C-terminal domain is a catalytic domain containing all methyltransferase motifs and is required for plasma membrane association. Vertebrate PRMT5 is usually depicted as a tetramer and to be complexed with its partner WD repeat protein MEP50, also known as WD Repeat Domain 77 (WDR77) or androgen co-activator p44, to form a heteroctomeric complex. This complex has a much higher methyltransferase activity than PRMT5 alone [8, 9].
PRMT5 is an epigenetic modifier that can regulate gene expression by methylating histones. Histone H2A has been reported to be methylated by PRMT5 on Arg3 as H2AR3me2s, while histone H3 can be methylated by PRMT5 on both Arg2 and Arg8 as H3R2me2s and H3R8me2s. PRMT5 can also methylate histone H4 on Arg3 as H4R3me2s (Fig. 1) [10,11,12]. By modifying these residues in histone tails, PRMT5 functionally activates or represses gene transcription and expression. For example, PRMT5, together with MEP50, has been found to methylate H2AR3 (H2AR3me2s) to repress gene expression and inhibit embryonic stem cell differentiation [13]. PRMT5 can also regulate the function of breast cancer stem cells via recruitment to the forkhead box protein 1 (FOXP1) promoter, methylation of H3R2 (H3R2me2s), facilitation of H3K4me3 and activation of FOXP1 gene expression [14]. The symmetric dimethylation (me2) of H3R8 and H4R3 has been shown to result in both transcriptional repression and activation. PRMT5 has been reported to interact with specificity protein 1 (SP1) to form a transcriptional repressor complex and to silence miR-29b via H4R3 methylation (H4R3me2) [15]. PRMT5 has also been found to increase the methylation of H3R8 (H3R8me2s) and H4R3 (H4R3me2s) in lymphoid cancer cells and to repress the expression of suppressor of tumorigenicity 7 (ST7), thereby promoting lymphoid cancer cell proliferation [11, 16]. In colorectal cancer cells, H4R3me2s and H3R8me2s methylation levels were found to be reduced upon PRMT5 knockdown, resulting in decreased FGFR3 and eIF4E transcription and expression [17].

Major PRMT5 methylated substrates. PRMT5 is a type II arginine methyltransferase that catalyzes monomethylarginine (MMA) and symmetric dimethylarginine (SDMA) formation. Most substrates that are methylated by PRMT5 can be divided into two major groups. One group contains histone proteins, including H3R2 (activates gene transcription), H2AR3 (represses gene transcription), as well as H3R8 and H4R3 (activate or repress gene transcription). The other group contains non-histone proteins that can regulate different cell functions, such as transcription (spliceosome Sm proteins, nucleoplasmin and nucleolin), proliferation (EGFR, FEN1 and RAF), apoptosis (p53), metabolism (SREBP1), cellular integrity (MBP and GM130), genome stability (Piwi proteins) and immune responses (HOXA9, BCL6 and NF-κB/p65)
PRMT5 can also methylate non-histone proteins and thereby regulate cellular processes, including transcription, proliferation, apoptosis, metabolism, cellular integrity, genome stability and immune responses (Fig. 1) [18]. PRMT5, as a component of the methylosome complex, acts as the enzymatic machinery. MEP50 is an adaptor molecule in the methylosome complex that connects PRMT5 to its substrates. This methylosome is crucial for spliceosome assembly and activity and directly methylates multiple components of the splicing machinery to regulate RNA splicing [19]. PRMT5 also methylates the well-known tumor suppressor protein p53 to regulate cell proliferation, cell cycle progression and cell death [20]. Furthermore, B-cell lymphoma 6 protein (BCL6) methylation via PRMT5 has been found to be necessary for germinal center formation and cell survival in lymphoma [21]. Recently, some novel PRMT5 targets have been identified by global proteomics profiling, which may refine our knowledge of the function of PRMT5 [22].
3 Regulation of PRMT5 is critical in cancer
Over the last 50 years, ample studies have consistently shown PRMT5 to be an oncoprotein that regulates a range of important cellular processes involved in cancer development via cross-talk with several signaling pathways [23, 24]. The majority of these studies focused on the function of PRMT5 in different cancers and addressed how PRMT5 can promote cancer cell proliferation, whereas others found that PRMT5 can also regulate other aspects of cancer development (Fig. 2).

PRMT5 functions in cancer cells and the tumor microenvironment. PRMT5 functions as a key regulator of cancer development. The expression levels of PRMT5 are regulated by microRNAs, and its overexpression in cancer cells has been found to improve cell survival, proliferation, migration and metabolism, and to inhibit cell apoptosis. PRMT5 is also required for cancer stem cell survival, self-renewal and differentiation. In addition, in the tumor microenvironment, PRMT5 inhibition in Treg cells causes downregulation of Treg inhibitory functions
3.1 Regulation of cancer cell survival, proliferation and apoptosis
In cancer cells, alternative mRNA processing may alter proteomic diversity, leading to dysregulated cellular processes [25]. PRMT5 plays important roles in mRNA splicing by methylating spliceosomal (Sm) proteins, such as D1, D3 and B/B’ [26]. In B-cell lymphoma, PRMT5 transcription was found to be upregulated by the MYC gene, and to be required for maintaining splicing fidelity, since it could methylate Sm proteins. PRMT5 deletion ex vivo has been found to lead to alternative splicing, reduced tumor cell viability, increased apoptosis, G1 cell cycle arrest and decreased tumor cell proliferation [27]. In glioblastoma, PRMT5 knockdown or inhibition was found to impair detained intron (DI) splicing, which led to downregulated proliferation-related gene transcription, resulting in tumor cell growth defects [28]. In breast cancer, PRMT5 along with WDR77 symmetrically dimethylates zinc finger protein 326 (ZNF326). Loss of PRMT5 and methylation of ZNF326 generates defects in alternative splicing of many genes that regulate cell proliferation and, finally, inhibit tumor cell proliferation [29]. In several hematologic and solid tumors, use of the PRMT5 inhibitor GSK3326595 has been found to attenuate tumor cell growth and survival. Many methylated PRMT5 substrates have been identified using mass-spectrometric analysis, including those involved in RNA splicing and processing. In addition, RNA-sequencing (RNA-Seq) has revealed induction of PRMT5 inhibition via alternative splicing of murine double minute 4 (MDM4), which can subsequently activate the well-known tumor suppressor p53 [30].
Repair of DNA damage ensures genome integrity. Mutation or dysregulation of genes that regulate DNA repair are commonly observed in cancer, resulting in uncontrolled cell proliferation [31]. In the peritumoral skin of melanoma, overexpression of PRMT5 and other genomic regulators has been observed. In addition, overexpression of genes that regulate histone dimethylation, cell cycle progression, DNA repair and nuclear protein import has been observed in melanoma, indicating that epigenetic modifications in peritumoral skin may be involved in melanoma development [32]. A potential mechanism could be that PRMT5 functions as a key regulator of homologous recombination (HR)-mediated double-strand break (DSB) repair. Since PRMT5 has been shown to symmetrically dimethylate RuvB-like 1 protein (RUVBL1), it may trigger acetylation of H4K16 by Tat-interactive protein-60KDa (TIP60) and facilitate p53 binding protein1 (53BP1) displacement from DSBs [33]. Moreover, in response to camptothecin, PRMT5 can directly bind tyrosyl-DNA phosphodiesterase 1 (TDP1) and dimethylate it, thus stimulating TDP1 repair function and promoting cell survival [34]. In acute myeloid leukemia (AML) cells, PRMT5 inhibition correlates with DNA damage, and its depletion has been found to cause DNA damage accumulation and cell cycle arrest. The reason behind this could be that PRMT5 silencing induces alternative splicing of histone-modification and DNA-repair factor TIP60/KAT5, leading to impaired HR-DNA repair [35]. Though data explaining the mechanism underlying the association between PRMT5 and DNA repair in cancer are scarce, together these results indicate that protein methylation, regulated by PRMT5, plays an important role in DNA repair processes.
DNA repair and cell cycle progression are tightly connected, i.e., p53 activated by DNA damage can cause cell cycle arrest and, ultimately, apoptosis. Since p53 is a major substrate of PRMT5 methylation, PRMT5 may regulate p53-mediated cell cycle arrest and apoptosis [20]. Another factor that promotes cell cycle progression and induces apoptosis is E2F transcription factor 1 (E2F-1), which has also been found to be directly methylated by PRMT5 [36]. In cancer, PRMT5 may also methylate Kruppel-like factor 4 (KLF4), leading to KLF4 ubiquitination inhibition. Elevated KLF4 protein levels lead to p21 upregulation, Bcl-2-associated X (BAX) downregulation, cancer cell genome stability impairment, increased cancer cell growth and survival and, finally, cancer progression [37]. In hepatocellular carcinoma cells, PRMT5 has been found to induce ERK phosphorylation to downregulate BTG2, whereas overexpression of B-cell translocation gene 2 (BTG2) in these cells caused cell cycle arrest in the G1 phase [38]. In lymphoma, PRMT5 has been found to promote cell cycle progression and proliferation by regulating the WNT/β-catenin and AKT/GSK3β pathways through increased transcription of their downstream target genes, including cyclin D1, c-MYC and survivin [39]. In glioblastoma, PRMT5 activity has been found to be required for the internal ribosome entry site activity of cyclin D1 and c-MYC via a heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1)-dependent pathway. When using a mTOR inhibitor to treat glioblastoma, PRMT5 activity was found to be upregulated, indicating a potential of using both mTOR and PRMT5 inhibitors to treat glioblastoma [40]. Similarly, in melanoma combinations of cyclin-dependent kinase 4/6 (CDK4/6) and PRMT5 inhibitors may provide an efficient therapeutic strategy, since in CDK4/6 inhibitor-resistant melanoma cells PRMT5 inhibition sensitivity is increased. This increase is dependent on MDM4, since PRMT5 regulates the splicing of MDM4 mRNA and MDM4 is a negative regulator of p53, which in turn regulates the cell cycle [41].
One characteristic of cancer cells is excessive proliferation, caused by evasion of apoptosis. Several reports have shown PRMT5 to play vital roles in this process. As mentioned earlier, E2F-1 is a substrate of PRMT5, and the PRMT5-E2F-1 pathway has been found to regulate Dicentrarchus labrax fucose-binding lectin (DlFBL)- and Strongylocentrotus purpuratus rhamnose-binding lectin (SpRBL)-induced cancer cell apoptosis [42]. Programmed cell death protein 4 (PDCD4) has been reported to promote apoptosis. In breast cancer, PRMT5 methylates PDCD4 and promotes PDCD4 expression. High expression of both PRMT5 and PDCD4 correlates with a worse outcome for patients with breast cancer [43]. Arginine methyltransferase inhibitor 1 (AMI-1), a PRMT5 inhibitor, has been found to strongly inhibit colorectal cancer growth in a mouse model via H4R3me2s and H3R8me2s methylation of fibroblast growth factor receptor 3 (FGFR3) and eukaryotic translation initiation factor 4E (eIF4E) gene promoters. AIM-1-induced cancer cell apoptosis via an increased BAX/Bcl-2 ratio was also observed in the same mouse model [17]. In bladder cancer, it has been found that PRMT5 enhances NF-κB activation, inhibits the expression of the anti-apoptotic B-cell lymphoma-extra large (BCLXL) and baculoviral IAP repeat-containing protein 1 (cIAP1) genes and, in addition, decreases cancer cell death [44].
3.2 Regulation of cancer cell migration
Cancer is difficult to cure, primarily due to metastasis. Previous studies have shown that PRMT5 can repress SNAIL-dependent gene expression, such as that of E-cadherin [45]. PRMT5 mRNA levels have also been found to correlate with lymph node metastasis in patients with gastric cancer, and in a gastric cancer cell line PRMT5 knockdown inhibited its proliferation, invasion and migration [46]. PRMT5 was also found to regulate epithelial-mesenchymal transition (EMT) and esophageal cancer cell migration [47]. More recently, PRMT5 has been reported to be guided by Cbp/p300-interacting transactivator 2 (CITED2) to nucleolin, thereby promoting prostate cancer cell migration through EMT induction [48]. Also, in prostate cancer miR-1266 has been found to target PRMT5 and, thereby, to inhibit cancer cell growth and metastasis [49]. Though most of the studies regarding PRMT5 in cancer cell migration have been performed in vitro, it is very well possible that PRMT5 may regulate cancer cell migration in vivo through EMT-correlated gene expression alterations via histone methylation. The exact role of PRMT5 in cancer metastasis needs, however, further confirmation and the underlying mechanisms need to be resolved to finally prevent cancer metastasis.
3.3 Regulation of cancer cell metabolism
Considering their high proliferative capability, cancer cells need to acquire nutrients from the tumor microenvironment while competing with normal cells. A common feature of cancer cells is the reprogramming of their metabolism, including metabolic changes in glucose, amino acids and fatty acids [50]. Through a phenomenon called the Warburg effect, cancer cells increase their rates of aerobic glycolysis, increase the rate of glucose uptake and secrete high levels of lactic acid [51]. Pioneering work showed that PRMT5 methylates H3R2me2, thereby enhancing chromatin accessibility to promote CREB phosphorylation, and increasing hepatic glucose production and gluconeogenic gene expression [52]. Further work in hepatocellular carcinoma has shown glucose induction to increase PRMT5 and CDK4 binding, activate CDK4-pRB-E2F-mediated transcription and promote cell proliferation and tumor growth. A recent study has shown that PRMT5 knockdown in a pancreatic cancer cell model reduced glucose intake, as well as lactate levels. 18F-fluorodeoxyglucose (18F-FDG) measurements by PET/CT scans have shown that patients with higher PRMT5 expression levels exhibit enhanced glycolytic capacity. In a xenograft mouse model, PRMT5 silencing concordantly decreased 18F-FDG uptake. Mechanistically, PRMT5 was found to inhibit F-Box and WD repeat domain containing 7 (FBW7) via the suppression of FBW7 gene promoter activity, leading to increased c-Myc expression and enhanced aerobic glycolysis in pancreatic cancer cells [53].
Due to the rapid proliferation of cancer cells, there is an increased requirement of amino acids [54]. Methylthioadenosine phosphorylase (MTAP) is an enzyme acting in the methionine salvage pathway that converts methylthioadenosine (MTA) back to methionine and adenine. In a variety of tumors, MTAP has been found to be lost due to deletion of the MTAP gene or methylation of its promoter. In 2016, two reports showed that in MTAP-deleted cancer cells, MTA is accumulated, leading to reduced PRMT5 enzymatic activity. Furthermore, these MTAP-deficient cancer cells were selectively sensitized during PRMT5 inhibition, thus indicating that PRMT5 inhibitors are potentially applicable to drug development for the treatment of MTAP-deficient tumors [55, 56]. Not long thereafter, researchers found this vulnerability to extend to decreases in MAT2A (a PRMT5 substrate) and RIOK1 (a PRMT5 interacting protein), although it could not increase the sensitivity to RIOK1 inhibition alone [57, 58]. A wider range of experiments was subsequently performed in more cancer cell lines, and the results showed that MTAP deficiency may also serve as a predictive marker for the ability of PRMT inhibitors to treat tumors, since PRMT5 inhibitors were found to synergize with PRMT1 inhibitors to inhibit tumor cell growth [59].
Tumor cells show altered lipid metabolism that reactivates de-novo lipid synthesis. They also show a strong lipid and cholesterol avidity. Currently, higher lipid droplet and stored-cholesteryl ester contents have become hallmarks of cancer aggressiveness [60]. In tumors, one of the factors triggering de-novo lipogenesis is the excessive activation of sterol regulatory element-binding proteins (SREBPs), which can upregulate lipogenic enzymes. PRMT5 symmetrically dimethylates SREBP1a at R321 and prevents the phosphorylation of SREBP1a at S430, subsequently leading to its disassociation from FBW7 to avoid protein degradation, so that stabilized SREBP1a can increase de-novo lipogenesis and promote cancer cell growth [61]. In addition, in clear cell renal cell carcinoma, PRMT5 was found to interact with LINC01138, thereby regulating SREBP1 methylation and protein stability [62]. Together, these data indicate that PRMT5 may be important for the regulation of glucose, amino acid and fatty acid metabolic pathways in cancer.
3.4 Regulation of cancer stem cell survival, self-renewal and differentiation
Another reason as to why cancer is difficult to cure is the existence of cancer stem cells (CSCs), which have the ability to differentiate into all types of cells in the tumor, and self-renew and sustain for a long time. Several reports have shown that PRMT5 activity regulates CSC survival, self-renewal and differentiation. In chronic myelogenous leukemia (CML), the survival and self-renewal capacities of leukemic stem cells is one of the causes of therapeutic resistance. Silencing PRMT5 or blocking PRMT5 activity disrupts this ability by epigenetically inhibiting dishevelled segment polarity protein 3 (DVL3) expression and downregulating Wnt/β-catenin signaling [63]. In solid tumors such as breast cancer, PRMT5 has been found to facilitate H3R2me2s methylation and to activate FOXP1 expression to regulate the proliferation and self-renewal of breast cancer stem cells [14]. In glioblastoma, PRMT5 has been found to be required for the self-renewal ability of undifferentiated stem cell-like populations of cells [64]. In hepatocellular carcinoma, PRMT5 has been found to promote the self-renewal and differentiation abilities of cancer stem cells by facilitating H4R3me2s methylation and repressing HNF4α gene expression [65]. In line with this, loss of PRMT5 function is known to be embryonically lethal, since PRMT5 methylates H2AR3me2s, represses differentiation-related gene expression and, finally, inhibits embryonic development [13]. In adults, loss of PRMT5 leads to rapid hematopoietic stem cell exhaustion via apoptosis, which is triggered by PRMT5-induced p53 activity [66]. Although the PRMT5 methylation targets are different in these processes, functional loss of PRMT5 inhibits CSC activity, suggesting the possibility that when PRMT5 inhibitors are used to treat cancer, they may be effective since they can target both cancer cells and CSCs.
3.5 Regulation of immune cell function
The tumor immune microenvironment not only includes tumor cells and stromal cells, but also many kinds of immune cells, such as T-cells, natural killer cells (NK cells), B-cells, macrophages, neutrophils and others. Immunotherapies that have recently shown positive results in cancer treatment are those that help the immune system to recognize and kill tumor cells. Due to the complexity and diversity of the tumor microenvironment, immunotherapy may not always lead to a positive response [67]. To date, there is only one report clearly showing that a PRMT5 inhibitor may be applicable to immunotherapy based on a mouse model. The authors of this study found that peripheral regulatory T-cells (Tregs) exhibit limited suppressive functions in a Treg-specific PRMT5 knock-out (KO) mouse model and that a PRMT5 inhibitor could reduce human Treg functions. Mechanistically, PRMT5 has been found to symmetrically dimethylate FOXP3 at R48/R51 and inhibit Treg suppressive functions, thereby reducing breast cancer tumor growth [68]. PRMT5 has also been found to regulate CD4+ T-cell expansion and to promote IL-2 production [69]. T-cell-specific deletion of PRMT5 causes a decrease in the number of peripheral CD4+ and CD8+ T-cells and in the number of thymic NK cells [70]. Recently, another group also showed T-cell survival and proliferation, of which the maintenance by cytokine signaling was dependent on PRMT5 expression [71]. As yet, no T-cell-specific PRMT5 KO tumor model has been tested but, based on current findings, we predict that PRMT5 silencing in T-cells may promote tumor growth. As the ratios of T-cell infiltration are diverse in different tumors, studies regarding the use of PRMT5 inhibitors should clarify their potential as drugs in different tumor models.
3.6 Regulation of PRMT5 expression by microRNAs
PRMT5 methylates histone or non-histone proteins to promote cancer. In cancer cells, the PRMT5 expression level has been found to be regulated by several microRNAs. MicroRNAs are small non-coding RNAs that regulate gene expression via RNA silencing or post-transcriptional regulation. They have been confirmed to play prominent roles in tumor development and progression [72]. In B-cell lymphoma, downregulation of miR-92b and miR-96 has been found to enhance the translation of PRMT5 mRNA and to increase PRMT5 protein expression [16]. In another lymphoma-related study, PRMT5-specific microRNAs 19a, 25, 32, 92, 92b and 96 were found to upregulate PRMT5 expression in the tumor cells [73]. In glioma, PRMT5 has been found to act as a miR-4518 target [74]. Interestingly, in B-cell lymphoma, PRMT5 was found to enhance the expression of itself by repressing miR-96 via a PRMT5/p65/HDAC3 complex feedback loop [75]. Further studies are recommended to explore whether these miRNAs, or other miRNAs that downregulate PRMT5 expression, may serve as therapeutic targets.
4 PRMT5 inhibitors and (pre-)clinical trials
PRMT5−/− mice show early embryonic lethality at E6.5, conditional PRMT5 knockout (cKO) in Treg cells causes severe scurfy-like autoimmunity, and PRMT5 cKO in neurons causes balance disorders, tremors, akinesis and hypomyelination. In a model of experimental hematology, PRMT5 cKO was found to cause aberrant erythroid differentiation and T-cell development. In a model to study development and germ cell fate, PRMT5 cKO caused disruption of primordial germ cell formation [12, 13, 68, 76,77,78]. All these phenotypes indicate that PRMT5 is an important factor during normal development and homeostasis. Moreover, PRMT5 has often been found to be highly expressed in tumors, with oncogene-like properties, owing to its ability to repress tumor suppressor gene expression. Therefore, several PRMT5 inhibitors have been used to explore potential cancer therapy approaches (Table 1).
The first PRMT5-specific compound, EPZ015666 (also known as GSK3235025), was reported in 2015 to treat a model of mantle cell lymphoma (MCL). EPZ015666, which is an orally administered inhibitor, showed significant and dose-dependent antitumor activity in xenograft MCL models [79]. To date, several PRMT5 inhibitors (non-specific), including EPZ004777, DS-437, LLY-283, CMP5, HLCL-61 and PR5-LL-CM01, have been used to treat different types of tumors. Some of the inhibitors exhibited high IC50 values and may, therefore, not be suited for future application. Some, however, have shown higher anti-tumor efficacies than EPZ015666, such as for example PR5-LL-CM01 in both pancreatic ductal adenocarcinoma and colorectal carcinoma. EPZ015666 binds to a complex composed of PRMT5, MEP50 and S-adenosylmethionine (SAM). DS-437, LLY-283, EPZ004777 and CMP5 are SAM competitive inhibitors. HLCL-61 and PR5-LL-CM01 were designed in silico to bind to the PRMT5 catalytic site, although not functioning as SAM-competitive inhibitors. Some of these inhibitors have only been tested in vitro in tumor cell line models and not in vivo yet. Overall, the modes of action of the different PRMT5 inhibitors still need to be better understood, and their efficacies to be pursued and verified in vivo.
Based on advances in the design of inhibitors and pre-clinical studies using mouse models, three PRMT5 inhibitors (GSK3326595, JNJ-64619178 and PF-06939999) are currently being used in four clinical trials (Table 2) for the treatment of both hematologic and solid tumors, including primary and metastatic tumors.
GSK3326595 is a substrate competitive inhibitor of PRMT5. There are two clinical trials going on of this inhibitor, and both of them are in phase I, tested as oral treatments. The purpose of the NCT03614728 study is to evaluate the safety, tolerability and clinical activity of this inhibitor in patients with relapsed and refractory myelodysplastic syndrome, chronic myelomonocytic leukemia and hypo-proliferative AML. If the result of this evaluation meets the pre-specified criteria, this clinical trial will be expanded to a phase II evaluation of this inhibitor. Another study of the PRMT5 inhibitor GSK3326595 (NCT02783300) aims to evaluate the safety, pharmacokinetics, pharmacodynamics and clinical activity of the inhibitor for patients with advanced or recurrent solid tumors and those with selected solid tumors and non-Hodgkin’s lymphomas. In this clinical trial, pembrolizumab will be used together with GSK3326595 for some patients. GSK3326595 has been found to inhibit tumor cell growth both in vivo and in vitro. Siu et al. showed that GSK3326595 monotherapy is active for relapsed and refractory solid tumors. In addition, GSK3326595 monotherapy has been found to significantly inhibit SDMA levels in patient plasmas and tumors. Clinical responses were observed at 200 mg QD (four times a day), and 400 mg QD was selected for dose expansion based on safety, efficacy, pharmacokinetic (PK), and pharmacodynamic (PD) data. Adverse events were common for patients receiving GSK3326595, but were effectively manageable with dose modifications, and even reversible with dose interruption. In adenoid cystic carcinoma, GSK3326595 treatment resulted in durable partial responses and prolonged stable disease [85, 86].
The PRMT5 inhibitor JNJ-64619178 is orally active and also in a phase I clinical trial. The purpose of this study is (1) to identify the maximum-tolerated dose in participants with relapsed/refractory B-cell non-Hodgkin’s lymphoma or advanced solid tumors, (2) to identify the recommended phase II dose(s) for non-Hodgkin’s lymphoma and other advanced solid tumors and (3) to evaluate inhibitor tolerability in participants with lower risk myelodysplastic syndromes. JNJ-64619178 has high potency, oral PK and safety properties. As its mode of action, it binds to the SAM- and protein substrate-binding pockets of the PRMT5/MEP50 complex. JNJ-64619178 has been found to inhibit growth in many different cancer cell lines and in mouse xenograft models of human non-small cell lung cancer and small cell lung cancer [87,88,89,90].
PF-06939999 is also in phase I for oral administration. This study was started in 2019 and aims to evaluate the safety, PKs and PDs in subjects with advanced or metastatic non-small cell lung cancer, head and neck squamous cell carcinoma, esophageal cancer, endometrial cancer, cervical cancer and bladder cancer. PF-06939999 is an inhibitor with potential inhibitory effects on proliferative and neoplastic activities.
The status of these ongoing clinical trials and preliminary data give us some hints for considering when and how to use these PRMT5 inhibitors. (1) Different PRMT5 inhibitors may be suitable for different cancer cell types. In lung cancer, for example, JNJ-64619178 may serve as a suitable selective PRMT5 inhibitor. Whether all the different inhibitors have specificities for certain cancer types needs further exploration. (2) PRMT5 depletion is known to be embryonic lethal and, thus, lack of selectivity of PRMT5 inhibitors in cancer and other cell types may induce toxicity later in clinical trials, as shown by the preliminary results using GSK3326595. (3) To date, little is known about how PRMT5 inhibitors affect immune cells in the tumor microenvironment and how this will affect the human immune system. This could be a potential source of side effects. (4) Resistance is a serious threat to targeted therapies, as PRMT5 has been shown to regulate proliferation and self-renewal in some types of cancer stem cells. Clinical trial results may help to evaluate the therapeutic benefit for recurrence rates. Since PRMT5 plays crucial roles in both healthy and cancer states, better designs of PRMT5 inhibitors are required to selectively reduce cancer-associated high PRMT5 activities, with minimal side effects. Thus, PRMT5 may be a viable target for therapeutic strategies, provided that clinical trials obtain encouraging (efficacy, tolerability) results.
5 Conclusions and perspectives
PRMT5 methylates histone or non-histone proteins to regulate gene expression, promote cancer cell proliferation and migration, activate or repress signal transduction, regulate cancer cell metabolism, and promote the self-renewal and differentiation abilities of cancer stem cells. PRMT5 inhibitors may add to the efficacy of immunotherapy. However, why PRMT5 overexpression elicits different effects in different tumor types still needs to be resolved. Additional mouse or cell line models exhibiting PRMT5 up- or downregulation need to be studied, and more PRMT5 substrates to be discovered. Since PRMT5 regulation occurs in either the nucleus or the cytoplasm, it is difficult to use PRMT5 expression as a biomarker to predict cancer status before obtaining tumor tissue. Technical tools for drug development are evolving rapidly, and exploratory studies on specific PRMT5 inhibitors are improving. Also, mechanistic studies of the processes underlying the therapeutic potentials of PRMT5 inhibitors will be a promising area of research. Several PRMT5 inhibitors have shown positive results in mouse models and are currently tested in clinical trials. PRMT5 inhibitors may show promise for cancer treatment either alone, or in combination with other currently used or future therapeutic modalities. Finally, PRMT5 inhibitors may show benefit for people with tumors that do not respond to checkpoint inhibitor therapy. Through their concerted actions on both tumor cells and tumor microenvironmental components, PRMT5 inhibitors may turn out to be highly effective in cancer therapy.
Data availability
Not applicable.
References
H.P. Mohammad, O. Barbash, C.L. Creasy, Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat. Med. 25, 403–418 (2019)
G.S. Baldwin, P.R. Carnegie, Specific enzymic methylation of an arginine in the experimental allergic encephalomyelitis protein from human myelin. Science 171, 579–581 (1971)
S. Brostoff, E.H. Eylar, Localization of methylated arginine in the A1 protein from myelin. Proc. Natl. Acad. Sci. U. S. A. 68, 765–769 (1971)
J. Jarrold, C.C. Davies, PRMTs and arginine methylation: Cancer’s best-kept secret? Trends Mol. Med. 25, 993–1009 (2019)
N. Stopa, J.E. Krebs, D. Shechter, The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell. Mol. Life Sci. 72, 2041–2059 (2015)
X.J. Ma, Q. Lu, M. Grunstein, A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in Saccharomyces cerevisiae. Genes Dev. 10, 1327–1340 (1996)
B.P. Pollack, S.V. Kotenko, W. He, L.S. Izotova, B.L. Barnoski, S. Pestka, The human homologue of the yeast proteins Skb1 and Hsl7p interacts with Jak kinases and contains protein methyltransferase activity. J. Biol. Chem. 274, 31531–31542 (1999)
S. Antonysamy, Z. Bonday, R.M. Campbell, B. Doyle, Z. Druzina, T. Gheyi, B. Han, L.N. Jungheim, Y. Qian, C. Rauch, M. Russell, J.M. Sauder, S.R. Wasserman, K. Weichert, F.S. Willard, A. Zhang, S. Emtage, Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U. S. A. 109, 17960–17965 (2012)
M.C. Ho, C. Wilczek, J.B. Bonanno, L. Xing, J. Seznec, T. Matsui, L.G. Carter, T. Onikubo, P.R. Kumar, M.K. Chan, M. Brenowitz, R.H. Cheng, U. Reimer, S.C. Almo, D. Shechter, Structure of the arginine methyltransferase PRMT5-MEP50 reveals a mechanism for substrate specificity. PLoS One 8, e57008 (2013)
A. Di Lorenzo, M.T. Bedford, Histone arginine methylation. FEBS Lett. 585, 2024–2031 (2011)
S.K. Kota, C. Roening, N. Patel, S.B. Kota, R. Baron, PRMT5 inhibition promotes osteogenic differentiation of mesenchymal stromal cells and represses basal interferon stimulated gene expression. Bone 117, 37–46 (2018)
A. Scaglione, J. Patzig, J. Liang, R. Frawley, J. Bok, A. Mela, C. Yattah, J. Zhang, S.X. Teo, T. Zhou, S. Chen, E. Bernstein, P. Canoll, E. Guccione, P. Casaccia, PRMT5-mediated regulation of developmental myelination. Nat. Commun. 9, 2840 (2018)
W.W. Tee, M. Pardo, T.W. Theunissen, L. Yu, J.S. Choudhary, P. Hajkova, M.A. Surani, Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 24, 2772–2777 (2010)
K. Chiang, A.E. Zielinska, A.M. Shaaban, M.P. Sanchez-Bailon, J. Jarrold, T.L. Clarke, J. Zhang, A. Francis, L.J. Jones, S. Smith, O. Barbash, E. Guccione, G. Farnie, M.J. Smalley, C.C. Davies, PRMT5 is a critical regulator of breast cancer stem cell function via histone methylation and FOXP1 expression. Cell Rep. 21, 3498–3513 (2017)
S.S. Tarighat, R. Santhanam, D. Frankhouser, H.S. Radomska, H. Lai, M. Anghelina, H. Wang, X. Huang, L. Alinari, A. Walker, M.A. Caligiuri, C.M. Croce, L. Li, R. Garzon, C. Li, R.A. Baiocchi, G. Marcucci, The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 30, 789–799 (2016)
S. Pal, R.A. Baiocchi, J.C. Byrd, M.R. Grever, S.T. Jacob, S. Sif, Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 26, 3558–3569 (2007)
B. Zhang, S. Dong, R. Zhu, C. Hu, J. Hou, Y. Li, Q. Zhao, X. Shao, Q. Bu, H. Li, Y. Wu, X. Cen, Y. Zhao, Targeting protein arginine methyltransferase 5 inhibits colorectal cancer growth by decreasing arginine methylation of eIF4E and FGFR3. Oncotarget 6, 22799–22811 (2015)
R.S. Blanc, S. Richard, Arginine methylation: The coming of age. Mol. Cell 65, 8–24 (2017)
J.Y. Fong, L. Pignata, P.A. Goy, K.C. Kawabata, S.C. Lee, C.M. Koh, D. Musiani, E. Massignani, A.G. Kotini, A. Penson, C.M. Wun, Y. Shen, M. Schwarz, D.H. Low, A. Rialdi, M. Ki, H. Wollmann, S. Mzoughi, F. Gay, C. Thompson, T. Hart, O. Barbash, G.M. Luciani, M.M. Szewczyk, B.J. Wouters, R. Delwel, E.P. Papapetrou, D. Barsyte-Lovejoy, C.H. Arrowsmith, M.D. Minden, J. Jin, A. Melnick, T. Bonaldi, O. Abdel-Wahab, E. Guccione, Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 36, 194-209 e9 (2019)
M. Jansson, S.T. Durant, E.C. Cho, S. Sheahan, M. Edelmann, B. Kessler, N.B. La Thangue, Arginine methylation regulates the p53 response. Nat. Cell Biol. 10, 1431–1439 (2008)
X. Lu, T.M. Fernando, C. Lossos, N. Yusufova, F. Liu, L. Fontan, M. Durant, H. Geng, J. Melnick, Y. Luo, F. Vega, V. Moy, G. Inghirami, S. Nimer, A.M. Melnick, I.S. Lossos, PRMT5 interacts with the BCL6 oncoprotein and is required for germinal center formation and lymphoma cell survival. Blood 132, 2026–2039 (2018)
D. Musiani, J. Bok, E. Massignani, L. Wu, T. Tabaglio, M.R. Ippolito, A. Cuomo, U. Ozbek, H. Zorgati, U. Ghoshdastider, R.C. Robinson, E. Guccione, T. Bonaldi, Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal. 12, eaat8388 (2019)
H. Shailesh, Z.Z. Zakaria, R. Baiocchi, S. Sif, Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 9, 36705–36718 (2018)
Y. Yang, M.T. Bedford, Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50 (2013)
P.P. Coltri, M.G.P. Dos Santos, G. H. G. da Silva, Splicing and cancer: Challenges and opportunities. Wiley Interdiscip. Rev. RNA 10, e1527 (2019)
A.B. Prusty, R. Meduri, B.K. Prusty, J. Vanselow, A. Schlosser, U. Fischer, Impaired spliceosomal UsnRNP assembly leads to Sm mRNA down-regulation and Sm protein degradation. J. Cell Biol. 216, 2391–2407 (2017)
C.M. Koh, M. Bezzi, D.H. Low, W.X. Ang, S.X. Teo, F.P. Gay, M. Al-Haddawi, S.Y. Tan, M. Osato, A. Sabo, B. Amati, K.B. Wee, E. Guccione, MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100 (2015)
C.J. Braun, M. Stanciu, P.L. Boutz, J.C. Patterson, D. Calligaris, F. Higuchi, R. Neupane, S. Fenoglio, D.P. Cahill, H. Wakimoto, N.Y.R. Agar, M.B. Yaffe, P.A. Sharp, M.T. Hemann, J.A. Lees, Coordinated splicing of regulatory detained introns within oncogenic transcripts creates an exploitable vulnerability in malignant glioma. Cancer Cell 32, 411-426 e11 (2017)
M. Rengasamy, F. Zhang, A. Vashisht, W.M. Song, F. Aguilo, Y. Sun, S. Li, W. Zhang, B. Zhang, J.A. Wohlschlegel, M.J. Walsh, The PRMT5/WDR77 complex regulates alternative splicing through ZNF326 in breast cancer. Nucleic Acids Res. 45, 11106–11120 (2017)
S.V. Gerhart, W.A. Kellner, C. Thompson, M.B. Pappalardi, X.P. Zhang, R. Montes de Oca, E. Penebre, K. Duncan, A. Boriack-Sjodin, B. Le, C. Majer, M.T. McCabe, C. Carpenter, N. Johnson, R.G. Kruger, O. Barbash, Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep 8, 9711 (2018)
B.M. Lorton, D. Shechter, Cellular consequences of arginine methylation. Cell. Mol. Life Sci. 76, 2933–2956 (2019)
A. Uzdensky, S. Demyanenko, M. Bibov, S. Sharifulina, O. Kit, Y. Przhedetski, V. Pozdnyakova, Expression of proteins involved in epigenetic regulation in human cutaneous melanoma and peritumoral skin. Tumour Biol. 35, 8225–8233 (2014)
T.L. Clarke, M.P. Sanchez-Bailon, K. Chiang, J.J. Reynolds, J. Herrero-Ruiz, T.M. Bandeiras, P.M. Matias, S.L. Maslen, J.M. Skehel, G.S. Stewart, C.C. Davies, PRMT5-dependent methylation of the TIP60 coactivator RUVBL1 is a key regulator of homologous recombination. Mol. Cell 65, 900-916 e7 (2017)
I. Rehman, S.M. Basu, S.K. Das, S. Bhattacharjee, A. Ghosh, Y. Pommier, B.B. Das, PRMT5-mediated arginine methylation of TDP1 for the repair of topoisomerase I covalent complexes. Nucleic Acids Res. 46, 5601–5617 (2018)
P.J. Hamard, G.E. Santiago, F. Liu, D.L. Karl, C. Martinez, N. Man, A.K. Mookhtiar, S. Duffort, S. Greenblatt, R.E. Verdun, S.D. Nimer, PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell. Rep. 24, 2643–2657 (2018)
E.C. Cho, S. Zheng, S. Munro, G. Liu, S.M. Carr, J. Moehlenbrink, Y.C. Lu, L. Stimson, O. Khan, R. Konietzny, J. McGouran, A.S. Coutts, B. Kessler, D.J. Kerr, N.B. Thangue, Arginine methylation controls growth regulation by E2F-1. EMBO J. 31, 1785–1797 (2012)
D. Hu, M. Gur, Z. Zhou, A. Gamper, M.C. Hung, N. Fujita, L. Lan, I. Bahar, Y. Wan, Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 6, 8419 (2015)
H. Jiang, Y. Zhu, Z. Zhou, J. Xu, S. Jin, K. Xu, H. Zhang, Q. Sun, J. Wang, J. Xu, PRMT5 promotes cell proliferation by inhibiting BTG2 expression via the ERK signaling pathway in hepatocellular carcinoma. Cancer Med. 7, 869–882 (2018)
J. Chung, V. Karkhanis, R.A. Baiocchi, S. Sif, Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/beta-catenin and AKT/GSK3beta proliferative signaling. J. Biol. Chem. 294, 7692–7710 (2019)
B. Holmes, A. Benavides-Serrato, J.T. Saunders, K.A. Landon, A.J. Schreck, R.N. Nishimura, J. Gera, The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J. Neurooncol. 145, 11–22 (2019)
S. AbuHammad, C. Cullinane, C. Martin, Z. Bacolas, T. Ward, H. Chen, A. Slater, K. Ardley, L. Kirby, K.T. Chan, N. Brajanovski, L.K. Smith, A.D. Rao, E.J. Lelliott, M. Kleinschmidt, I.A. Vergara, A.T. Papenfuss, P. Lau, P. Ghosh, S. Haupt, Y. Haupt, E. Sanij, G. Poortinga, R.B. Pearson, H. Falk, D.J. Curtis, P. Stupple, M. Devlin, I. Street, M.A. Davies, G.A. McArthur, K.E. Sheppard, Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc. Natl. Acad. Sci. U. S. A. 116, 17990–18000 (2019)
L. Wu, X. Yang, X. Duan, L. Cui, G. Li, Exogenous expression of marine lectins DlFBL and SpRBL induces cancer cell apoptosis possibly through PRMT5-E2F-1 pathway. Sci. Rep. 4, 4505 (2014)
M.A. Powers, M.M. Fay, R.E. Factor, A.L. Welm, K.S. Ullman, Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 71, 5579–5587 (2011)
G. Hu, X. Wang, Y. Han, P. Wang, Protein arginine methyltransferase 5 promotes bladder cancer growth through inhibiting NF-kB dependent apoptosis. EXCLI J. 17, 1157–1166 (2018)
Z. Hou, H. Peng, K. Ayyanathan, K.P. Yan, E.M. Langer, G.D. Longmore, F.J. Rauscher III, The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol. Cell. Biol. 28, 3198–3207 (2008).
M. Kanda, D. Shimizu, T. Fujii, H. Tanaka, M. Shibata, N. Iwata, M. Hayashi, D. Kobayashi, C. Tanaka, S. Yamada, G. Nakayama, H. Sugimoto, M. Koike, M. Fujiwara, Y. Kodera, Protein arginine methyltransferase 5 is associated with malignant phenotype and peritoneal metastasis in gastric cancer. Int. J. Oncol. 49, 1195–1202 (2016)
J. Zhang, Q. Liu, L. Qiao, P. Hu, G. Deng, N. Liang, J. Xie, H. Luo, J. Zhang, Novel role of granulocyte-macrophage colony-stimulating factor: antitumor effects through inhibition of epithelial-to-mesenchymal transition in esophageal cancer. Onco Targets Ther. 10, 2227–2237 (2017)
S.H. Shin, G.Y. Lee, M. Lee, J. Kang, H.W. Shin, Y.S. Chun, J.W. Park, Aberrant expression of CITED2 promotes prostate cancer metastasis by activating the nucleolin-AKT pathway. Nat. Commun. 9, 4113 (2018)
C.M. Sun, G.M. Zhang, H.N. Qian, S.J. Cheng, M. Wang, M. Liu, D. Li, MiR-1266 suppresses the growth and metastasis of prostate cancer via targeting PRMT5. Eur. Rev. Med. Pharmacol. Sci. 23, 6436–6444 (2019)
X. Li, M. Wenes, P. Romero, S.C. Huang, S.M. Fendt, P.C. Ho, Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 16, 425–441 (2019)
M.G. Vander Heiden, L.C. Cantley, C.B. Thompson, Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009)
W.W. Tsai, S. Niessen, N. Goebel, J.R. Yates 3rd., E. Guccione, M. Montminy, PRMT5 modulates the metabolic response to fasting signals. Proc. Natl. Acad. Sci. U. S. A. 110, 8870–8875 (2013)
Y. Qin, Q. Hu, J. Xu, S. Ji, W. Dai, W. Liu, W. Xu, Q. Sun, Z. Zhang, Q. Ni, B. Zhang, X. Yu, X. Xu, PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell. Commun. Signal. 17, 30 (2019)
M.J. Lukey, W.P. Katt, R.A. Cerione, Targeting amino acid metabolism for cancer therapy. Drug Discov. Today. 22, 796–804 (2017)
G.V. Kryukov, F.H. Wilson, J.R. Ruth, J. Paulk, A. Tsherniak, S.E. Marlow, F. Vazquez, B.A. Weir, M.E. Fitzgerald, M. Tanaka, C.M. Bielski, J.M. Scott, C. Dennis, G.S. Cowley, J.S. Boehm, D.E. Root, T.R. Golub, C.B. Clish, J.E. Bradner, W.C. Hahn, L.A. Garraway, MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218 (2016)
K.J. Mavrakis, E.R. McDonald III, M.R. Schlabach, E. Billy, G.R. Hoffman, A. deWeck, D.A. Ruddy, K. Venkatesan, J. Yu, G. McAllister, M. Stump, R. deBeaumont, S. Ho, Y. Yue, Y. Liu, Y. Yan-Neale, G. Yang, F. Lin, H. Yin, H. Gao, D.R. Kipp, S. Zhao, J.T. McNamara, E.R. Sprague, B. Zheng, Y. Lin, Y.S. Cho, J. Gu, K. Crawford, D. Ciccone, A.C. Vitari, A. Lai, V. Capka, K. Hurov, J.A. Porter, J. Tallarico, C. Mickanin, E. Lees, R. Pagliarini, N. Keen, T. Schmelzle, F. Hofmann, F. Stegmeier, W.R. Sellers, Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213 (2016)
K. Marjon, M.J. Cameron, P. Quang, M.F. Clasquin, E. Mandley, K. Kunii, M. McVay, S. Choe, A. Kernytsky, S. Gross, Z. Konteatis, J. Murtie, M.L. Blake, J. Travins, M. Dorsch, S.A. Biller, K.M. Marks, MTAP deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 axis. Cell. Rep. 15, 574–587 (2016)
A. Hormann, B. Hopfgartner, T. Kocher, M. Corcokovic, T. Krammer, C. Reiser, G. Bader, J. Shi, K. Ehrenhofer, S. Wohrle, N. Schweifer, C.R. Vakoc, N. Kraut, M. Pearson, M. Petronczki, R.A. Neumuller, RIOK1 kinase activity is required for cell survival irrespective of MTAP status. Oncotarget 9, 28625–28637 (2018)
A. Fedoriw, S.R. Rajapurkar, S. O’Brien, S.V. Gerhart, L.H. Mitchell, N.D. Adams, N. Rioux, T. Lingaraj, S.A. Ribich, M.B. Pappalardi, N. Shah, J. Laraio, Y. Liu, M. Butticello, C.L. Carpenter, C. Creasy, S. Korenchuk, M.T. McCabe, C.F. McHugh, R. Nagarajan, C. Wagner, F. Zappacosta, R. Annan, N.O. Concha, R.A. Thomas, T.K. Hart, J.J. Smith, R.A. Copeland, M.P. Moyer, J. Campbell, K. Stickland, J. Mills, S. Jacques-O’Hagan, C. Allain, D. Johnston, A. Raimondi, M. Porter Scott, N. Waters, K. Swinger, A. Boriack-Sjodin, T. Riera, G. Shapiro, R. Chesworth, R.K. Prinjha, R.G. Kruger, O. Barbash, H.CR87P. Mohammad, Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 36, 100–114 e25 (2019)
S. Beloribi-Djefaflia, S. Vasseur, F. Guillaumond, Lipid metabolic reprogramming in cancer cells. Oncogenesis 5, e189 (2016)
L. Liu, X. Zhao, L. Zhao, J. Li, H. Yang, Z. Zhu, J. Liu, G. Huang, Arginine methylation of SREBP1a via PRMT5 promotes de novo lipogenesis and tumor growth. Cancer Res. 76, 1260–1272 (2016)
X. Zhang, J. Wu, C. Wu, W. Chen, R. Lin, Y. Zhou, X. Huang, The LINC01138 interacts with PRMT5 to promote SREBP1-mediated lipid desaturation and cell growth in clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 507, 337–342 (2018)
Y. Jin, J. Zhou, F. Xu, B. Jin, L. Cui, Y. Wang, X. Du, J. Li, P. Li, R. Ren, J. Pan, Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Invest. 126, 3961–3980 (2016)
Y.K. Banasavadi-Siddegowda, A.M. Welker, M. An, X. Yang, W. Zhou, G. Shi, J. Imitola, C. Li, S. Hsu, J. Wang, M. Phelps, J. Zhang, C.E. Beattie, R. Baiocchi, B. Kaur, PRMT5 as a druggable target for glioblastoma therapy. Neuro Oncol. 20, 753–763 (2018)
B.N. Zheng, C.H. Ding, S.J. Chen, K. Zhu, J. Shao, J. Feng, W.P. Xu, L.Y. Cai, C.P. Zhu, W. Duan, J. Ding, X. Zhang, C. Luo, W.F. Xie, Targeting PRMT5 activity inhibits the malignancy of hepatocellular carcinoma by promoting the transcription of HNF4alpha. Theranostics 9, 2606–2617 (2019)
D.Q. Tan, Y. Li, C. Yang, J. Li, S.H. Tan, D.W.L. Chin, A. Nakamura-Ishizu, H. Yang, T. Suda, PRMT5 Modulates splicing for genome integrity and preserves proteostasis of hematopoietic stem cells. Cell. Rep. 26, 2316-2328 e6 (2019)
M. Binnewies, E.W. Roberts, K. Kersten, V. Chan, D.F. Fearon, M. Merad, L.M. Coussens, D.I. Gabrilovich, S. Ostrand-Rosenberg, C.C. Hedrick, R.H. Vonderheide, M.J. Pittet, R.K. Jain, W. Zou, T.K. Howcroft, E.C. Woodhouse, R.A. Weinberg, M.F. Krummel, Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018)
Y. Nagai, M.Q. Ji, F. Zhu, Y. Xiao, Y. Tanaka, T. Kambayashi, S. Fujimoto, M.M. Goldberg, H. Zhang, B. Li, T. Ohtani, M.I. Greene, PRMT5 associates with the FOXP3 homomer and when disabled enhances targeted p185(erbB2/neu) tumor immunotherapy. Front. Immunol. 10, 174 (2019)
L.M. Webb, S.A. Amici, K.A. Jablonski, H. Savardekar, A.R. Panfil, L. Li, W. Zhou, K. Peine, V. Karkhanis, E.M. Bachelder, K.M. Ainslie, P.L. Green, C. Li, R.A. Baiocchi, M. Guerau-de-Arellano, PRMT5-selective inhibitors suppress inflammatory T cell responses and experimental autoimmune encephalomyelitis. J. Immunol. 198, 1439–1451 (2017)
M. Inoue, K. Okamoto, A. Terashima, T. Nitta, R. Muro, T. Negishi-Koga, T. Kitamura, T. Nakashima, H. Takayanagi, Arginine methylation controls the strength of gammac-family cytokine signaling in T cell maintenance. Nat. Immunol. 19, 1265–1276 (2018)
Y. Tanaka, Y. Nagai, M. Okumura, M.I. Greene, T. Kambayashi, PRMT5 is required for T cell survival and proliferation by maintaining cytokine signaling. Front. Immunol. 11, 621 (2020)
Y. Peng, C.M. Croce, The role of MicroRNAs in human cancer. Signal Transduct. Target Ther. 1, 15004 (2016)
L. Wang, S. Pal, S. Sif, Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 28, 6262–6277 (2008)
Y.F. Lu, X.L. Cai, Z.Z. Li, J. Lv, Y.A. Xiang, J.J. Chen, W.J. Chen, W.Y. Sun, X.M. Liu, J.B. Chen, LncRNA SNHG16 functions as an oncogene by sponging MiR-4518 and up-regulating PRMT5 expression in glioma. Cell. Physiol. Biochem. 45, 1975–1985 (2018)
L. Alinari, K.V. Mahasenan, F. Yan, V. Karkhanis, J.H. Chung, E.M. Smith, C. Quinion, P.L. Smith, L. Kim, J.T. Patton, R. Lapalombella, B. Yu, Y. Wu, S. Roy, A. De Leo, S. Pileri, C. Agostinelli, L. Ayers, J.E. Bradner, S. Chen-Kiang, O. Elemento, T. Motiwala, S. Majumder, J.C. Byrd, S. Jacob, S. Sif, C. Li, R.A. Baiocchi, Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 125, 2530–2543 (2015)
M. Bezzi, S.X. Teo, J. Muller, W.C. Mok, S.K. Sahu, L.A. Vardy, Z.Q. Bonday, E. Guccione, Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 27, 1903–1916 (2013)
Z. Li, J. Yu, L. Hosohama, K. Nee, S. Gkountela, S. Chaudhari, A.A. Cass, X. Xiao, A.T. Clark, The Sm protein methyltransferase PRMT5 is not required for primordial germ cell specification in mice. EMBO J. 34, 748–758 (2015)
F. Liu, G. Cheng, P.J. Hamard, S. Greenblatt, L. Wang, N. Man, F. Perna, H. Xu, M. Tadi, L. Luciani, S.D. Nimer, Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J. Clin. Invest. 125, 3532–3544 (2015)
E. Chan-Penebre, K.G. Kuplast, C.R. Majer, P.A. Boriack-Sjodin, T.J. Wigle, L.D. Johnston, N. Rioux, M.J. Munchhof, L. Jin, S.L. Jacques, K.A. West, T. Lingaraj, K. Stickland, S.A. Ribich, A. Raimondi, M.P. Scott, N.J. Waters, R.M. Pollock, J.J. Smith, O. Barbash, M. Pappalardi, T.F. Ho, K. Nurse, K.P. Oza, K.T. Gallagher, R. Kruger, M.P. Moyer, R.A. Copeland, R. Chesworth, K.W. Duncan, A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 11, 432–437 (2015)
S.R. Daigle, E.J. Olhava, C.A. Therkelsen, C.R. Majer, C.J. Sneeringer, J. Song, L.D. Johnston, M.P. Scott, J.J. Smith, Y. Xiao, L. Jin, K.W. Kuntz, R. Chesworth, M.P. Moyer, K.M. Bernt, J.C. Tseng, A.L. Kung, S.A. Armstrong, R.A. Copeland, V.M. Richon, R.M. Pollock, Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65 (2011)
D. Smil, M.S. Eram, F. Li, S. Kennedy, M.M. Szewczyk, P.J. Brown, D. Barsyte-Lovejoy, C.H. Arrowsmith, M. Vedadi, M. Schapira, Discovery of a dual PRMT5-PRMT7 inhibitor. ACS Med. Chem. Lett. 6, 408–412 (2015)
Z.Q. Bonday, G.S. Cortez, M.J. Grogan, S. Antonysamy, K. Weichert, W.P. Bocchinfuso, F. Li, S. Kennedy, B. Li, M.M. Mader, C.H. Arrowsmith, P.J. Brown, M.S. Eram, M.M. Szewczyk, D. Barsyte-Lovejoy, M. Vedadi, E. Guccione, R.M. Campbell, LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity. ACS Med. Chem. Lett. 9, 612–617 (2018)
K. Zhu, C.S. Jiang, J. Hu, X. Liu, X. Yan, H. Tao, C. Luo, H. Zhang, Interaction assessments of the first S-adenosylmethionine competitive inhibitor and the essential interacting partner methylosome protein 50 with protein arginine methyltransferase 5 by combined computational methods. Biochem. Biophys. Res. Commun. 495, 721–727 (2018)
L. Prabhu, H. Wei, L. Chen, O. Demir, G. Sandusky, E. Sun, J. Wang, J. Mo, L. Zeng, M. Fishel, A. Safa, R. Amaro, M. Korc, Z.Y. Zhang, T. Lu, Adapting AlphaLISA high throughput screen to discover a novel small-molecule inhibitor targeting protein arginine methyltransferase 5 in pancreatic and colorectal cancers. Oncotarget 8, 39963–39977 (2017)
D. Rasco, A. Tolcher, L.L. Siu, K. Heinhuis, S. Postel-Vinay, O. Barbash, J.L. Egger, S. Gorman, T. Horner, A. Dhar, B.E. Kremer, A phase I, open-label, dose-escalation study to investigate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of GSK3326595 in subjects with solid tumors and non-Hodgkin’s lymphoma. Cancer Res. 77, supplement (2017)
D.W. Rasco, L.L. Siu, S Postel Vinay, P Martin Romano, J. Menis, F.L. Opdam, K.M. Heinhuis, J.L. Egger, S.A. Gorman, R. Parasrampuria, K. Wang, B.E. Kremer, M.M. Gounder, METEOR-1: A phase I study of GSK3326595, a first-in-class protein arginine methyltransferase 5 (PRMT5) inhibitor, in advanced solid tumours. Ann. Oncol. 30, v159 (2019)
D. Brehmer, T.F. Wu, G. Mannens, L. Beke, P. Vinken, D. Gaffney, W.M. Sun, V. Pande, J.W. Thuring, H. Millar, I. Poggesi, I. Somers, A. Boeckx, M. Parade, E. van Heerde, T. Nys, C. Yanovich, B. Herkert, T. Verhulst, M. Du Jardin, L. Meerpoel, C. Moy, G. Diels, M. Viellevoye, W. Schepens, A. Poncelet, J.T. Linders, E.C. Lawson, J.P. Edwards, D. Chetty, S. Laquerre, M. V. Lorenzi, A novel PRMT5 inhibitor with potent in vitro and in vivo activity in preclinical lung cancer models. Cancer Res. 77, supplement (2017)
T.F. Wu, H. Millar, D. Gaffney, L. Beke, G. Mannens, P. Vinken, I. Sommers, J.W. Thuring, W.M. Sun, C. Moy, V. Pande, J.G. Zhou, N. Haddish-Berhane, M. Salvati, S. Laquerre, M.V. Lorenzi, D. Brehmer, JNJ-64619178, a selective and pseudo-irreversible PRMT5 inhibitor with potent in vitro and in vivo activity, demonstrated in several lung cancer models. Cancer Res. 78, supplement (2018)
X. Li, C. Wang, H. Jiang, C. Luo, A patent review of arginine methyltransferase inhibitors (2010–2018). Expert Opin. Ther. Pat. 29, 97–114 (2019)
H.J. Millar, D. Brehmer, T. Verhulst, N. Haddish-Berhane, T. Greway, D. Gaffney, A. Boeckx, E. Van Heerde, T. Nys, J. Portale, U. Philippar, T.F. Wu, S. Laquerre, K. Packman, In vivo efficacy and pharmacodynamic modulation of JNJ-64619178, a selective PRMT5 inhibitor, in human lung and hematologic preclinical models. Cancer Res. 79, supplement (2019)
Funding
This study was supported by grants from the National Key R&D Program of China (2019YFA0111000), the Shanghai Science and Technology Committee (20ZR1448900), the Shanghai Healthy Committee (202040121), the National Natural Science Foundation of China (No. 81671590) and the Innovative research team of high-level local universities in Shanghai.
Author information
Authors and Affiliations
Contributions
YY wrote the manuscript. HN provided direction and reviewed and revised the manuscript. Both authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Code availability
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yuan, Y., Nie, H. Protein arginine methyltransferase 5: a potential cancer therapeutic target. Cell Oncol. 44, 33–44 (2021). https://doi.org/10.1007/s13402-020-00577-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-020-00577-7