
Abstract
Purpose
Due to a lack of effective targeted therapies, patients with metastatic triple-negative breast cancer (TNBC) have poor clinical outcomes. Epithelial to mesenchymal transition (EMT) is known to contribute to cancer progression, invasiveness and multidrug resistance (MDR). There is a strong correlation between various drug efflux mechanisms, cancer stem cells and tumor microenvironments, which in turn is synchronized by complex signaling crosstalk between EMT and MDR. We hypothesize that combining these regulatory connections with targeted combinatorial therapies may be an effective approach to annihilate the progression/metastasis of TNBC.
Methods
AlamarBlue assays were used to depict TNBC cell viability, whereas flow cytometry was used to detect apoptotic cell populations, reactive-oxygen species (ROS) levels as well as mitochondrial depolarization. qRT-PCR, Western blotting and confocal microscopy were used to provide molecular-level information of the genes and proteins involved.
Results
Our initial analyses showed that targeting EGFR by either erlotinib (EGFR inhibitor) or lapatinib (EGFR/HER-2 inhibitor) alone was ineffective against TNBC. Interestingly, we subsequently found that a low dose of lapatinib did act as a substrate rather than as an inhibitor facilitating EMT and MDR, leading to metastasis. Additional gene expression studies indicated that co-targeting the EGFR and Wnt/β-catenin pathways with lapatinib and XAV939 (a tankyrase inhibitor) promoted mesenchymal to epithelial transition (MET). Application of these inhibitors led to a 5.62-fold increase in the epithelial marker E-cadherin and a 3.33-fold decrease in the stemness marker EpCAM, with concomitant 1.5-fold and 3.22-fold reductions in the ABC transporters ABCB1 and ABCG2, respectively. These co-targeting effects resulted in overcoming EMT and MDR, which in turn was highlighted by reduced levels of pEGFR, pAKT, pMAPK, pSTAT-3, pGSK-3β and β-catenin.
Conclusions
Our data indicate that the synergistic action of targeting both the EGFR and Wnt/β-catenin signaling pathways in TNBC cells may open up new avenues for combatting this disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Epithelial to mesenchymal transition (EMT) is a complex molecular process by which epithelial cells lose their differentiated characteristics and instead gain mesenchymal features like increased motility, invasiveness and elevated resistance to apoptosis [1]. EMT plays a pivotal role in the morphogenesis of multicellular organisms, particularly during embryonic development and wound healing [2]. Inappropriate EMT induction may have, however, deleterious effects due to a compromised mechanical and physiological integrity of tissues, which in case of cancer may lead to metastasis. At the molecular level, cells undergoing EMT exhibit a decreased expression level of epithelial genes (i.e., E-cadherin, occludin, ZO-1) and an increased expression level of mesenchymal genes (i.e., N-cadherin, vimentin, fibronectin) [3]. Loss of E-cadherin is considered to be a hallmark of EMT [4]. Several crucial transcription factors regulate the processes underlying EMT (e.g., TWIST, SNAI1, SNAI2, ZEB1/2), which act in a concerted fashion with epigenetic mechanisms as well as post-translational protein modifications [5]. Although EMT is a process associated with normal development, it is increasingly being recognized as one of the central mechanisms responsible for the induction of invasive and metastatic properties in solid tumors.
The heterogeneity and plasticity of EMT phenotypes are varied and diverse. Apart from metastasis, it has also been found to regulate multidrug resistance (MDR). The link between EMT and MDR lies in a small population of cells known as cancer stem cells (CSCs), which are thought to be responsible for tumorigenesis [6]. Remarkably, it has been found that several of the signaling pathways activated during EMT and those driving CSCs are similar, including the Wnt, Hedgehog and Notch signaling pathways [6]. Earlier reports suggest that cells undergoing EMT exhibit stem cell-like properties, thus sharing key signaling pathways and MDR properties with CSCs [7]. CSCs confer MDR by excessive drug efflux conducted by multiple cell membrane transporter proteins, especially the ATP-binding cassette (ABC) transporter family of proteins [8].
EMT, invasiveness, drug resistance and metastasis are critical features of the most aggressive subtype of breast cancer (BC), known as triple-negative breast cancer (TNBC), which represents 15 to 20% of all BC [9]. Typical characteristics of TNBC are lack of expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER-2). TNBC occurs predominantly in premenopausal women [10]. Primarily due to a lack of effective targeted therapies, patients with metastatic TNBC (mTNBC) have a poor clinical outcome with a median overall survival (OS) time of approximately 13–16 months [11].
Genetic and immunohistochemical analyses have shown that the epidermal growth factor receptor (EGFR) is frequently overexpressed in TNBC, often correlating with a poor prognosis [12]. It is well-known that anomalous activation of EGFR in cancer cells results in deregulated proliferation, EMT, invasion, metastasis and resistance to apoptosis [12]. Clinical trials of EGFR inhibiting drugs as single agents have, however, shown ineffectiveness against TNBC, suggesting that EGFR does not represent a significant vulnerability [13]. Lapatinib, a dual tyrosine kinase inhibitor (EGFR/HER-2), was subsequently developed to target both receptors synergistically. Lapatinib alone, however, also lacks efficacy in treating TNBC.
Wnt/β-catenin signaling has been found to be another prominent contributor to EMT in various cancer types and to be highly dysregulated in TNBC. Earlier studies on TNBC indicate that EMT, cell migration, chemoresistance, colony formation and stem cell-like features are promoted by nuclear accumulation of β-catenin. This, in turn, emphasizes a pivotal role of canonical Wnt signaling as one of the driving forces of TNBC tumorigenesis and metastasis [14]. An intriguing correlation between EGFR and Wnt signaling is that both of them can activate β-catenin and thus induce EMT. Transactivation of β-catenin via EGFR-mediated PI3K/Akt activation suggests β-catenin activation via receptor tyrosine kinase pathways [15]. Inhibition of β-catenin may be instrumental in restraining EMT and drug resistance. Tankyrase 1 (TNKS1/PARP5A)-mediated poly-ADP-ribosylation activity degrades AXIN and thus triggers disruption of the β-catenin destruction complex. XAV939, a potent inhibitor of TNKS1, stabilizes AXIN, which in turn reduces β-catenin levels [16].
Given the strong implication of EGFR and Wnt/β-catenin signaling in the molecular pathogenesis of TNBC through EMT and MDR, we hypothesized that an effective combination of inhibitors, one directed against EGFR (lapatinib) and the other directed against TNSK1 (XAV939), may be a strategy to accomplish profound therapeutic effects in TNBC. We explored the effect of the inhibitor combination on TNBC cells by analyzing molecular changes in EMT markers in both monolayer cultures and multicellular tumor spheroids, which mimic in vivo conditions. The expression levels of genes contributing to EMT and MDR were found to be significantly reduced. The invasive nature and migratory potential of the TNBC cells were also found to be downregulated several-folds. As expected, we found that lapatinib showed more cytotoxicity than the EGFR-only targeting drug erlotinib. Subsequent treatment with lapatinib and XAV939 resulted in enhanced cytotoxicity. Importantly, this combination was found to show synergism and to be effective at very low doses compared to either therapy alone. The potential of co-targeting EGFR and Wnt/β-catenin signaling pathway components for a more efficacious TNBC treatment is highlighted.
2 Materials and methods
2.1 Cell lines and culture conditions
The human breast cancer cell lines MCF-7, MDA-MB-231 and MDA-MB-468 were purchased from the National Centre for Cell Science, Pune, India. They were cultured in Dulbecco’s Modified Eagle’s Medium-high glucose supplemented with L-glutamine, sodium pyruvate (Sigma-Aldrich, St. Louis, MI, USA), 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA), Sodium bicarbonate (Sigma-Aldrich), 100 units/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific). The cells were maintained at 37 °C in humidified air containing 5% CO2. To induce EMT, cells were starved for 4 h in serum-free medium and incubated in 20 nM EGF for 30 min prior to treatment.
2.2 Cell viability and drug combination assays
Cell viability was evaluated using an alamarBlue (Thermo Fisher Scientific) assay. Resazurin, the active ingredient of alamarBlue, is converted to its reduced form resorufin when entering into living cells [17]. Following treatment for 48 h of monolayer cultures in 96-well plates, 10 µl alamarBlue was directly added to the wells and incubated for 2 h at 37 °C under 5% CO2 humidified conditions. In case of spheroids, following treatment for 72 h, 20 µl alamarBlue was added and incubated for 4 h at 37 °C under 5% CO2 humidified conditions. After the respective incubations, absorbance was measured at 570 nm with a reference of 600 nm using a microplate reader (Infinite M200 Pro, Tecan, Switzerland). Cell viability (%) was determined using the following formula:
The inhibitory concentration-50 (IC50) values were determined from sigmoidal-dose response curves using GraphPad software. The combined effect of lapatinib and XAV939 was determined by analyzing the Chou and Talalay combination index (CI) using Calcusyn software (Biosoft) [18].
2.3 Cell cycle analysis
To analyze the effect of the drug combination on cell cycle progression, propidium iodide (PI) (Sigma Aldrich)-based flow cytometry analysis was performed. Cells were seeded at a density of 1 × 105 cells per 60 mm tissue culture petri-dish and allowed to attach for 12 h. Cells were synchronized prior to treatment by serum starvation for 36 h. Treatment with the inhibitors and their combination was performed for 48 h in complete media. Following treatment, cells were trypsinized, centrifuged at 650 rcf for 6 min and fixed by adding 70% chilled ethanol dropwise. Fixed cells were stored in -20 °C until further analysis as described by Riccardi et al. [19]. Briefly, cells were washed with chilled phosphate buffered saline (PBS) and incubated with RNAse for 1 h at 37 °C. Next, the cells were incubated with PI and stored in the dark on ice until further analysis. The samples were analyzed using a BD FacsCalibur, and the collected data were analyzed using FCS Express software.
2.4 Western blotting
Following treatment, total protein was extracted from the cells using RIPA lysis buffer (Sigma Aldrich). Protein from each sample was quantified, and equal amounts were subjected to SDS-PAGE and subsequently transferred to PVDF membranes. Next, the membranes were blocked using blocking buffer (4% BSA in TBST) and incubated overnight with primary antibodies. After washing and incubation with secondary antibodies, the signals were developed using chemiluminescent reagent (Sigma Aldrich), and images were acquired using ChemiDoc (BioRad). The images were quantified using ImageJ software. Anti-EGFR, anti-phospho-EGFR (Tyr1068), anti-Akt, anti-phospho-Akt (Ser473), anti-p44/42 MAPK (Erk1/2), anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), anti-Stat-3, anti-phospho-Stat-3 (Tyr705), anti-GSK-3β, anti-phospho- GSK-3β (Ser9), anti-E-cadherin, anti-N-cadherin and anti-Vimentin monoclonal antibodies were purchased from Cell Signaling Technology. Anti-β-catenin monoclonal antibody, anti-Rabbit IgG and anti-mouse IgG secondary antibodies were purchased from Sigma Aldrich. The antibodies and respective catalog numbers are listed in Supplementary Table S1.
2.5 Generation of spheroids
Spheroids of MCF-7, MDA-MB-231 and MDA-MB-468 cells were generated using a forced floating method [20, 21]. Cells were cultured as monolayers up to confluency, trypsinized and resuspended in DMEM. In the meantime, wells of 96-well plates were coated with agarose (1.5% w/v) containing serum-free DMEM. Next, the cells were seeded at a density of 2 × 104 cells/well, after which the plates were centrifuged at 700 rcf for 10 min. Subsequently, the 96-well plates were incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 96 h. The resulting spheroids were visually monitored with a Nikon Eclipse Ti microscope and used for subsequent experiments.
2.6 Quantitative real-time PCR
Following treatment, RNA was isolated from the cells using a RNeasy Mini kit (Qiagen). From the obtained RNA, cDNA was synthesized using an iScript cDNA synthesis kit (BioRad) according to the manufacturer’s instructions. Amplification of the cDNA of interest was performed using a PowerUp™ SYBR™ Green Master Mix (Applied Biosystems) and a Rotor-Gene Q (Qiagen) real-time PCR cycler. The data obtained were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and quantified using the delta-delta CT method [22]. The sequences of the primers used are listed in Supplementary Table S2. The obtained quantitative RT-PCR (qRT-PCR) data were analyzed using LinReg PCR software.
2.7 Apoptosis assay
Early apoptotic, apoptotic and necrotic cell populations were estimated using a FITC Annexin V Apoptosis Detection Kit (BD Biosciences). Following treatment, samples were processed according to the manufacturer’s instructions and analyzed using a CytoFLEX (Beckman Coulter) flow cytometer. Data analysis and fluorescence compensation were performed using CytoFLEX software.
2.8 Reactive oxygen species (ROS) detection assay
Cellular ROS generation following drug treatment was analyzed using 2’,7’-dichlorofluorescein diacetate (DCFDA) dye (Sigma Aldrich). DCFDA is a cell-permeant reagent, and in the presence of hydroxyl, peroxyl and other ROS activities within the cell, it is oxidized to 2’,7’-dichlorofluorescein (DCF), which gives a green fluorescence. For ROS detection, cells were incubated with 10 µM DCFDA for 30 min and treated with individual inhibitors and their combination for 6 h, after which the cells were trypsinized and analyzed using flow cytometry.
2.9 Mitochondrial membrane potential detection assay
Mitochondrial membrane potential was determined using JC-1 staining (Thermo Fisher Scientific). Cells were treated with inhibitors and their combination for 48 h. After completion of the treatment, JC-1 dye (10 µM) was added to each cell culture well for 20 min. Following incubation, the cells were trypsinized and analyzed using flow cytometry. Fluorescence intensities were collected in the red and green channels.
2.10 Live-dead cell imaging and immunofluorescence analysis
For live-dead cell imaging following drug treatment, spheroids were washed thrice with PBS and incubated with calcein-AM and PI to final concentrations of 2 µM and 4 µM, respectively. After incubation, the spheroids were washed thrice with PBS and imaged using a Zeiss LSM 880 confocal microscope in conjunction with Z-stacking analysis.
For immunofluorescence analysis, following drug treatment, cells were washed thrice with PBS, fixed with 4% formaldehyde for 15 min and rewashed. Next, the cells were immersed in blocking buffer for 2 h and subsequently incubated overnight at 4 °C with primary antibodies. After a second wash, the cells were incubated with a fluorochrome-conjugated secondary antibody for 2 h at room temperature in the dark. Next, the cells were rewashed and incubated with DAPI (1 µM) for 5 min. After a final wash, imaging was performed using a Zeiss LSM 880 confocal microscope.
2.11 Scratch wound-healing migration assay
Scratch wound-healing assays were performed using a protocol described by Wu et al. [23]. Briefly, cells were grown to 60–70% confluence in complete DMEM. Next, the media were replaced by 1% serum media for 24 h. Confluent cell monolayers were scraped with a sterile pipette tip to create ‘wounds’. The wounded monolayers were washed with PBS to remove cell debris. Next, the cells were treated with inhibitors and their combination and incubated at 37 °C under humidified conditions and 5% CO2 for a period of 24 h. Images of the ‘wounds’ before and after treatment were captured using a Nikon Eclipse Ti microscope and analyzed using ImageJ software.
2.12 Matrigel invasion assay
Invasion assays were performed using a method described by Chen et al. [24]. Briefly, matrigel (Sigma Aldrich) was diluted to 1 mg/ml in serum-free medium. The solution was poured into upper Boyden chamber transwell inserts (Corning) and incubated at 37 °C overnight. After solidification, 2 × 105 cells in serum-free media were seeded into the upper chambers, whereas the lower chambers were filled with 750 µl DMEM containing 10% FBS. Next, the cells were incubated at 37 °C for 24 h, after which the chamber’s upper sides were wiped with a wet swab to remove the cells and washed thrice with PBS. The lower sides of the chambers were incubated in 4% formaldehyde for 15 min at 37 °C, washed thrice with PBS, stained with DAPI (1 µM), rewashed thrice with PBS, and analyzed using a Zeiss LSM 880 confocal microscope. The fold change in fluorescence intensity in the DAPI stained cells was assessed by confocal microscopy for the invaded cells. DAPI stains specifically dsDNA, avoiding other non-specific staining.
2.13 Statistical analysis
All statistical analyses were performed using GraphPad prism software. The experimental data are expressed as mean ± SEM. The one-way ANOVA test was used to assess possible correlations between groups. A p-value < 0.05 was considered statistically significant where * p < 0.05, ** p < 0.001, *** p < 0.001, **** p < 0.0001, respectively.
3 Results
3.1 Co-targeting EGFR and Wnt/β-catenin signaling synergistically inhibits TNBC cell survival
MDA-MB-231 cells were treated with lapatinib and XAV939 alone and in combination to determine their effect on cell viability. Using an alamarBlue assay on lapatinib treated monolayer cells, we observed a significant dose-dependent decrease in cell viability (Fig. 1a). A several-fold higher dose of lapatinib was required for the spheroids (Fig. 1g). On the other hand, a higher dose of XAV939 was required in both cases to obtain cell death (Fig. 1b and h). The combination of lapatinib with XAV939 was found to decrease TNBC cell viability in a dose-dependent manner at lower concentrations in both monolayers and spheroids (Fig. 1c and i). The combination effect on spheroids was significant at a lower dose but, surprisingly, not effective at a higher dose (Fig. 1i). Combination index (CI) calculation revealed a CI value of < 1 for both cases, indicating a dose-dependent synergistic interaction between lapatinib and XAV939 (Fig. 1e, f, k, and l). Erlotinib did not show any significant inhibitory effect in both monolayer cultures and spheroids (Fig. 1d and j). Similar to the data reported by Bao et al. [25], we found that only targeting EGFR in TNBC using erlotinib is ineffective (Fig. 1d and j). Administration of the inhibitors in combination yielded higher anti-proliferative effects than when administered alone (Fig. 1a, b, and c). In case of spheroids, a relatable finding was observed as well (Fig. 1g, h, and i) [26]. Significant correlations were also observed in other TNBC cells, i.e., MDA-MB-468 and luminal type MCF-7 cells (Supplementary Fig. S1 and S2).

Determination of viability of MDA-MB-231 monolayer cultures by alamarBlue assay upon treatment with inhibitors for 48 h. a, b, c and d refer to treatment with lapatinib, XAV939, 6 µM lapatinib + XAV939 and erlotinib, respectively. e Combination index and f dose-response curves obtained following combination treatment. Determination of viability of MDA-MB-231 spheroids by alamarBlue assay upon treatment with inhibitors for 72 h. g, h, i and j refer to treatment with lapatinib, XAV939, 30 µM lapatinib + XAV939 and erlotinib, respectively. k Combination index and (l) dose-effect curve obtained following combination treatment. Drug combinations with CIs < 1.0 were considered as synergistic. Results expressed as the mean ± SEM are based on at least three independent experiments. The statistical significance was assessed versus that of the untreated cells. The significance level was set at p < 0.05 (*), p < 0.01 (**), p < 0.001 (***) or p < 0.0001 (****)
Live-dead cell imaging of spheroids following treatment provided a clear visualization of the effect of these inhibitors (Fig. 2). Fluorescence intensities of the spheroids presented in Fig. 2 were quantified and are presented in Supplementary Table S3. The percentages of live-dead cells were analyzed from the quantified fluorescence intensities and are graphically represented in Supplementary Fig. S3. Similar results were also obtained using z-stack projection of live-dead cell staining of MCF-7 spheroids (Supplementary Fig. S4, Table S4 and Fig. S5). Moreover, we found that the co-treatment markedly induced disintegration of the spheroids (Fig. 5a). Taken together, these data indicate that modulation of EGFR and Wnt/β-catenin signaling synergistically inhibits TNBC cell survival.

Live-dead cell visualization of MDA-MB-231 spheroids using calcein-AM/propidium iodide (PI) dual staining. Green fluorescence by calcein-AM refers to live cells, whereas red fluorescence by PI refers to dead cells. a Untreated spheroids and (b), (c) and (d) spheroids treated for 72 h with 30 µM lapatinib, 25 µM XAV939 and 30 µM lapatinib + 25 µM XAV939, respectively. Scale bar: 250 µM. (e) Z-stack projections of MDA-MB-231 spheroids
3.2 Inhibition of EGFR and Wnt/β-catenin signaling results in G0/G1 cell cycle arrest
To better document the effect of EGFR and Wnt/β catenin inhibition on cell growth/survival, cell cycle progression was monitored following 48 h treatment in case of monolayer cultures and 72 h in case of spheroids. A G0/G1 phase cell cycle arrest was observed in both cases. The percentage of G0/G1 phase cells was increased to 16.53% in monolayers and 13% in spheroids (Fig. 3a and b). Cyclin-D1 and cyclin-dependent kinase-4 (CDK-4) are regulatory proteins responsible for driving cell cycle progression from the G1 to S phase [27]. Thus, we set out to analyze the expression levels of cyclin-D1 and CDK-4 using qRT-PCR to better understand the mechanism of action of the inhibitors. The expression levels of cyclin-D1 and CDK-4 were found to be down-regulated by 3.57-fold and 3.33-fold, respectively (Supplementary Fig. S6). Therefore, we conclude that modulation of the EGFR and Wnt/β-catenin signaling pathway ablates cyclin-D1 and CDK-4, leading to G0/G1 cell cycle arrest.

Evaluation of cell cycle profile of MDA-MB-231 (a) monolayer cultures and (b) spheroids. Mitochondrial membrane potential detection of MDA-MB-231 monolayer cells using JC-1 staining (c). Flow cytometric analysis of apoptotic populations probed by annexin-V-FITC PI assay following treatment in MDA-MB-231 (d) monolayer cultures and (e) spheroids. Cellular ROS detection using a DCFDA-based flow cytometric assay in MDA-MB-231 (f) monolayer cultures and (g) spheroids
3.3 Suppression of EGFR and Wnt/β-catenin signaling generates ROS, depolarizes mitochondrial membrane potential and induces apoptosis
In an attempt to explicate the mechanism underlying cell death, the generation of ROS and mitochondrial membrane integrity were examined. Cellular ROS levels were detected using 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA)-based flow cytometry [28]. Our flow cytometric analyses showed that lapatinib and XAV939 in combination generated a 3.3-fold increase in ROS compared to the respective untreated controls (Fig. 3f). The combined administration did, however, not increase ROS generation in spheroids (Fig. 3g). An elevated ROS level manifests its detrimental effect on the integrity of the mitochondrial membrane and its associated trans-membrane potential [29]. The effect of ROS accumulation on mitochondrial membrane integrity was assessed using JC-1-based flow cytometry. The cyanine dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimi-dazolylcarbocyanine iodide) forms red aggregates in polarized mitochondria, whereas it remains as a monomer in depolarized mitochondria [30]. We observed a prominent red fluorescence from healthy mitochondria and a ~ 37% increase in green fluorescence from treated cells, suggesting mitochondrial membrane depolarization (Fig. 3c).
Cells react rapidly to redox imbalance with a plethora of biological responses, including cell cycle-specific growth arrest, transcription-coupled repair of damaged DNA and apoptosis. Following the trail of ROS generation and mitochondrial membrane potential disruption, the percentage of apoptotic cells was examined after combination treatment. Using flow cytometry, we found that co-therapy increased apoptotic cell populations by approximately 41.19% and 20% in monolayer cultures and spheroids, respectively (Fig. 3d and e). Similar results were obtained with MDA-MB-468 and MCF-7 cells (Supplementary Fig. S7 and S8). The percentages of apoptotic cell populations following co-treatment are listed in supplementary Table S5.
3.4 Inactivation of EGFR and Wnt/β-catenin signaling induces mesenchymal-epithelial transition (MET)
In the EMT process, tumor-associated epithelial cells obtain mesenchymal features with reduced cell-cell contacts and an increased motility, which play critical roles in invasion and metastasis. Both EGFR and Wnt/β-catenin signaling are crucial in EMT. Therefore, qRT-PCR was performed to evaluate changes in the expression of epithelial and mesenchymal markers under treated conditions. Loss of E-cadherin-mediated adhesion characterizes the transition from benign lesions to invasive, metastatic cancers [31]. Following combination treatment, E-cadherin was found to be upregulated by 5.7-fold. XAV939 alone upregulated E-cadherin expression by 2.5-fold, whereas lapatinib was found to be ineffective (Fig. 4a). Vimentin and N-cadherin are major markers of EMT [32, 33]. Although co-therapy did not alter the vimentin and N-cadherin levels, lapatinib treatment increased their expression levels by 3.35-fold and 6.10-fold, respectively (Fig. 4a). The expression level of key EMT-related proteins such as E-cadherin, N-cadherin and vimentin, was carried out using Western blot analysis. E-cadherin was found to be upregulated by 1.73-fold following combination treatment (Supplementary Fig. S9), whereas, N-cadherin and vimentin were found to be downregulated by 3-fold and 2.18-fold, respectively (Supplementary Fig. S9). Remarkably, a 14.3-fold decrease in vimentin expression was observed in MDA-MB-231 spheroids following co-treatment (Supplementary Fig. S9).

Graphical representation of changes in gene expression levels following inhibitor treatment quantified by qRT-PCR analysis. a, c, e, g and i show gene expression levels obtained from MDA-MB-231 monolayer cultures. b, d, f, h and j show gene expression levels obtained from MDA-MB-231 spheroids. Results are expressed as mean relative gene expression level compared to GAPDH ± SEM of three independent experiments, with p < 0.05 (*), p < 0.01 (**), p < 0.001 (***) or p < 0.0001 (****)
Besides EMT markers, some important proteins and transcription factors, such as fibronectin, Ki-67, Twist and SNAI2, are also implicated in the process of EMT. Although no significant alterations were observed for the transcription factors Twist and SNAI2, EpCAM and Ki-67 were found to be downregulated by 3.3-fold and.2.9-fold after combination therapy, respectively (Fig. 4c and g). The combination effect was also examined in tumor spheroids by qRT-PCR. Unlike in the monolayer cells, vimentin expression was low, and E-cadherin expression was only marginally elevated, even after 72 h of treatment (Fig. 4b). However, other EMT markers such as N-cadherin and fibronectin, and Ki-67 were not found to be significantly altered after combination treatment (Fig. 4b and h). While combination treatment led to decreased Twist expression, lapatinib alone increased its expression by two-fold. SNAI2 was upregulated under both treatment conditions (Fig. 4d). Exogenous EpCAM expression in TNBC cells has been found to promote EMT, to induce a CSC-like phenotype and to enhance metastasis in vitro and in vivo [34]. We found that after combination treatment, EpCAM was downregulated by 3.30-fold in monolayers but remained unaltered in spheroids (Fig. 4g and h). Expression analysis of EMT-related proteins in two other TNBC cell lines, MDA-MB-468 and MCF-7, yielded similar EMT reversal results (Supplementary Fig. S10 and Table S6). E-cadherin was, however, found to be downregulated in MCF-7 cells.
MDM2 plays a critical role in controlling p53 and, thus, apoptosis. MDM2 acts as a cellular antagonist of p53 [35]. It has also been reported that MDM2 increases drug resistance through inducing EMT independent of p53 [36]. We found that co-targeting EGFR and Wnt/β-catenin led to a decreased MDM2 level both in monolayers (1.26-fold) and tumor spheroids (4.16-fold), which incurred cell death following co-treatment (Fig. 4i and j). It is well-known that caveolin-1 overexpression is correlated with migration, metastasis and drug resistance [37]. We found that co-treatment reduced the caveolin-1 expression level by 1.72-fold and 1.50-fold in monolayers and spheroids, respectively (Fig. 4i and j).
Together, these findings indicate that combined treatment with EGFR and Wnt/β catenin signaling inhibitors induces MET and concomitant reductions in the metastatic and invasive potentials of TNBC cells.
3.5 Co-targeting EGFR and Wnt/β-catenin signaling decreases TNBC cell migration and invasion
During the process of EMT, epithelial cells reverse their morphology into mesenchymal cells and gain increased abilities for migration and invasion, which could result in metastasis. Since both EGFR and Wnt/β-catenin signaling are implicated in the migration and invasion of TNBC cells, we next investigated the combined effect of lapatinib and XAV939 on the migration and invasion of MDA-MB-231 cells. Migration was assessed using a scratch wound-healing assay, where the migration rate of the cells towards the wound area (created using a scratch) was determined. We found that the cells incubated with lapatinib, XAV939 and in combination showed slower wound healing abilities than the untreated cells. The rate of cell migration was found to be lowest in the combination treatment (Fig. 5b). In addition a Boyden chamber-based assay, in which cells invade a layer of matrigel on top of a membrane, was used to monitor the invasive capacity of MDA-MB-231 cells. After 24 h, a 3-fold reduction in invasion capacity was observed in cells treated with the combination of inhibitors compared to untreated cells (Fig. 5d). Of note, the effect on migration and invasion was stronger upon co-treatment compared to treatment with each single agent, confirming the synergistic effect of the inhibitors (Fig. 5c and e). These data indicate that co-inhibition of EGFR and Wnt/β-catenin signaling reduces the migration and invasion capacities of TNBC cells.

a Visualization of the effect of inhibitors and their combination on MDA-MB-231 spheroids using phase-contrast microscopy. Combination treatment results in visible disintegration of spheroids. b Scratch wound-healing assays of MDA-MB-231 monolayer cultures. c Graphical representation of changes in wound healing capacity following inhibitor treatment and their combination with respect to untreated samples. d Boyden-chamber invasion assays of MDA-MB-231 monolayer cultures. e Graphical representation of changes in invasiveness following inhibitor treatment and their combination with respect to untreated samples
3.6 Suppression of EGFR and Wnt/β-catenin signaling alters the expression of ABC transporters in TNBC cells
Drug efflux transport systems have extensively been studied owing to the phenomenon of MDR, in which cancer cells become cross-resistant to multiple cytotoxic anticancer drugs. MDR often results from the overexpression of ABC transporters [38]. This prompted us to study alterations in the expression of ABC subfamily B member 1 (ABCB1; also called P-glycoprotein), ABC subfamily C member 1 (ABCC1; also called MDR-associated protein 1), ABC subfamily G member 2 (ABCG2; also called breast cancer resistance protein), all known to be involved in MDR, by qRT-PCR. In Fig. 4e and f the expression levels of the ABC transporters in MDA-MB-231 monolayer cultures and spheroids are depicted. We found that combination treatment led to decreased ABCB1 expression in both monolayers and spheroids. However, lapatinib treatment alone increased ABCB1 expression by 1.5-fold and 5.87-fold in monolayers and spheroids, respectively. After combination treatment, the expression of ABCG2 was found to be reduced significantly in monolayers, but to be slightly increased in spheroids. Although co-treatment did not alter ABCC1 expression, a higher expression was observed in XAV939 treated spheroids. Together, these data indicate that downregulation of EGFR and Wnt/β-catenin signaling alters the expression of major MDR-associated genes in TNBC cells.
3.7 Alteration of EGFR and Wnt/β-catenin signaling downregulates pro-survival signals and reveals extensive crosstalk and downstream convergence
The effect of the inhibitors on the expression of pro-survival proteins was evaluated by Western blot analysis (Fig. 7a). EGF-treated MDA-MB-231 cells were incubated with varying concentrations of the inhibitors. We found that individual treatment with lapatinib or XAV939 reduced the pEGFR levels drastically, inferring possible crosstalk. Co-administration of lapatinib and XAV939 also reduced the phosphorylated receptor level (1.88-fold). At the indicated concentrations, each inhibitor was found to be insufficient to alter the level of pMAPK. Their combination, however, reduced the pMAPK level significantly. The activated form of AKT (pAKT) was found to be reduced more efficiently by XAV939 than by lapatinib, whereas the combination effectively diminished its level (3.125-fold). Like pMAPK, the inhibitor(s) alone could only marginally alter the level of β-catenin, but markedly (2.17-fold) when administered in combination. The PI3K-AKT signaling pathway is known to phosphorylate GSK-3β at Ser9, which leads to inactivation of GSK-3β and augmentation of β-catenin-TCF/LEF-1 transcriptional activity [39]. We found that the level of pGSK-3β was reduced significantly after combination treatment. Lapatinib alone reduced the p-GSK-3β level only marginally, whereas XAV939 had a better effect (2.22-fold). This finding is consistent with the previous results of the reduced pAKT level. pSTAT-3, another kinase with an interplaying role between the EGFR and Wnt/β-catenin signaling pathways, was also found to be downregulated (2.32-fold) after combination treatment.
Next, protein expression studies were carried out in spheroids to understand the effect of the inhibitors on cellular signaling. We found that the individual inhibitors and their combination showed almost similar effects in spheroids compared to monolayers (Fig. 7b). However, unlike in monolayers, XAV939 failed to show a significant effect on the pEGFR level in spheroids. The combination treatment was also not effective in altering the pSTAT-3 level.
Following EGF binding, EGFR is incorporated into clathrin-coated pits for further degradation in lysosomes [40]. Although endosomal EGF-EGFR complexes may retain cell signaling ability, lapatinib renders them inactive [41]. To understand whether co-treatment in the presence of EGF alters the surface expression of EGFR, immunofluorescence microscopic analyses were performed. We found that co-treatment did not alter EGFR surface expression (Fig. 6a). Similarly, AKT distribution was found to be unaltered (Fig. 6b). In line with the above Western blot results, cytosolic β-catenin was found to be significantly reduced (Fig. 6c). The fold change of fluorescence intensity in Fig. 6 was quantified and is graphically presented in supplementary Fig. S11.

Confocal images of MDA-MB-231 monolayer cultures immune-stained with (a) anti-EGFR antibody, (b) anti-AKT antibody and (c) anti-β-catenin antibody, respectively, visualized by Alexa Flour-488. Nuclei were stained with DAPI. Upper row refers to untreated samples and lower row refers to samples treated with lapatinib + XAV939 (6 µM LAP + 50 µM XAV) for 48 h. Scale bar, 20 µM
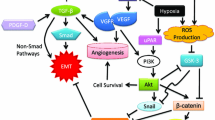
Our findings indicate crosstalk between the EGFR and Wnt/β-catenin pathways and their convergence to downstream signaling. Although a single inhibitor alone could influence the targets by blocking the phosphorylation of RTKs, the effects were only feebly percolated to its downstream effectors and unable to affect cell functions regulated by the targeted pathway. Therefore, blocking more than one pathway simultaneously may alter the effector functions (Figs. 7 and 8).

Representative Western blots showing EGFR/pEGFR, AKT/pAKT, MAPK/pMAPK, STAT-3/pSTAT-3, β-catenin/GSK-3β/pGSK-3β and β-actin levels in MDA-MB-231 cell extracts of (a) monolayer cultures and (b) spheroids. For monolayer cultures, cells were treated with inhibitors for 48 h, where the concentration of lapatinib is 6 µM and of XAV939 is 50 µM. Spheroids were treated using 30 µM lapatinib and 25 µM XAV939 for 72 h. β-actin serves as a loading control. Graphs represent the changes in the expression levels of pEGFR, pAKT, pMAPK, pSTAT-3 and pGSK-3β with respect to untreated samples following inhibitor treatment. The expression levels were deduced from the blots using ImageJ software

Schematic illustration of (a) overall observable cellular events and (b) details of intracellular signaling upon lapatinib-XAV939 co-treatment regulating EMT and MDR dynamics
4 Discussion
In TNBC, several signaling pathways have been found to be responsible for inducing EMT and maintaining CSC properties. Multiple crosstalks, feedback loops, and their essential roles in maintaining normal cellular developmental processes make it almost impossible to effectively target them and halt EMT progression. It is, therefore, quite perceptible that crosstalk between signaling pathways has to be obliterated in order to block EMT. To avert possible severe side effects arising from high concentrations of drugs in plasma, we examined a combination of small molecule inhibitors at low concentrations. We focused on two such inhibitors, lapatinib, an FDA approved drug for metastatic breast cancer [42] and XAV939, an inhibitor that is being tested at the preclinical stage [43]. It should be noted that although a few FDA-approved drugs do affect Wnt signaling, albeit nonspecifically, inhibitors explicitly targeting the Wnt/β-catenin pathway have only recently entered clinical trials [44]. We found that the combination effect on spheroids was significant at lower doses, but less effective at higher doses. This is possibly due to the complex ECM structure in spheroids that alters endosmosis. Cell-ECM and cell-cell interactions increase cell density and, thereby, may form a physical barrier that limits the penetration of compounds to cells, also known as limited mass transport effect [45]. At lower concentrations of the drug, endosmosis was possibly favored over higher concentrations. Also, increased interstitial fluid pressure (IFP) and hypoxic conditions in spheroids may attribute to the drug uptake process above a certain threshold [46].
Cyclin-D1 and CDK-4 partly mediate G1 to S-phase cell cycle progression by phosphorylation and inactivation of the retinoblastoma (Rb) protein, with a subsequent release of E2F transcription factors [27]. Tetsu et al. reported that β-catenin regulates the expression of Cyclin-D1 in colon carcinoma cells and that abnormal β-catenin expression levels may, therefore contribute to neoplastic transformation by causing Cyclin-D1 accumulation [47]. Cyclin-D1 is also known to be a downstream target of HER-2 and EGFR [48]. Evidently, we found that modulation of EGFR and Wnt/β-catenin signaling resulted in reduced expression of Cyclin-D1 and CDK-4, leading to G0/G1 cell cycle arrest. Similar to our findings, Masamha et al. reported that Cyclin-D1 degradation in ovarian cancer cells is sufficient to induce G0/G1 cell cycle arrest despite the constitutive expression of Cyclin-E2 [49]. The sub-G1 population, indicating cell shrinkage and DNA fragmentation, is considered to be a apoptotic cell population [50].
The effectiveness of traditional cancer chemotherapy is mostly based on the generation of ROS and, consequently, on increased oxidative stress that exceeds the reduction capacity of the cancerous tissues, ultimately leading to apoptosis or necrosis [51]. We found that lapatinib alone led to more ROS than the combination of inhibitors, which might be attributed to the activation of multiple death signaling cascades following combination therapy. Akin to our findings, Arid et al. reported that lapatinib may act as a potent inducer of ROS in inflammatory breast cancer (IBC) cells [52]. The cellular ROS levels were, however, found to be decreased following single or combined treatment of tumor spheroids. Increased ROS levels cause mitochondrial membrane hyper-polarization, which leads to the collapse of mitochondrial membrane potential (ΔΨm), mitochondrial translocation of Bax and Bad, cytochrome-c release and, ultimately, apoptosis [53]. Our JC-1 staining data revealed an increase in depolarized mitochondria, confirming ROS-mediated mitochondrial membrane damage after combination treatment. The cytotoxicity of an anticancer drug depends heavily on its ability to initiate apoptosis in the targeted cells. In this regard, lapatinib and XAV939 have both been reported to induce caspase-dependent apoptosis [54, 55]. Liu et al. reported an almost 40% increase in apoptosis of MDA-MB-231 cells after 72 h treatment with 10 µM lapatinib [56]. The observed 41.19% increase in apoptotic cell population after co-administration of lapatinib and XAV939 for 48 h at low concentrations substantiates the efficacy of the proposed treatment.
Considering the involvement of EGFR and Wnt/β-catenin signaling in inducing EMT and metastasis in TNBC, the effect of the inhibitor combination on EMT was evaluated. MDA-MB-231 cells are known to have an EMT-induced mesenchymal phenotype (Supplementary Fig. S12) [57]. Following combination treatment, downregulation of vimentin, N-cadherin, fibronectin, Ki-67, SNAI2, Twist and EpCAM, and upregulation of E-cadherin indicate a reversal of the EMT process, i.e., MET, which contributes to reduced motility, invasiveness and drug resistance. Strikingly, only lapatinib treatment induced the cells to express high levels of mesenchymal markers and reduced levels of epithelial markers. Previously Hsiao et al. also reported that lapatinib induced increased migratory and metastatic properties in MDA-MB-231 cells [58]. According to their findings, lapatinib induces IL-6 expression triggered through the MAPK pathway. Elevated expression of IL-6 is indeed known to contribute to enhanced cancer aggressiveness [59]. The here observed increased level of pMAPK after lapatinib treatment correlates with these studies and decodes the probable cause of the enhanced EMT markers. However, contrasting lapatinib results were obtained in spheroids with upregulated levels of E-cadherin and reduced levels of vimentin, N-cadherin and EpCAM. A plausible explanation for this phenomenon may be linked to the drug efflux transport system.
The major obstacle of successful chemotherapy treatment is multidrug resistance (MDR), predominantly due to overexpression of ATP-binding cassette (ABC) transporters [38]. Although ABCB1 and ABCG2 were found to be significantly reduced upon combination treatment, lapatinib alone increased the expression of ABCB1 significantly in both monolayers and spheroids. ABCG2 was, however, found to be reduced in monolayers and to be increased in spheroids. The drug efflux functions of ABCB1 and ABCG2 are linked to ATP hydrolysis, which is stimulated in the presence of the substrates. Polli et al. found that lapatinib acts both as a substrate and an inhibitor of ABCB1 and ABCG2 (IC50 values of 3.9 and 0.025 µmol/L, respectively) [60]. It was evident that lapatinib acted as a substrate for these two transporters at lower concentrations and as an inhibitor at higher concentrations. Based on higher IC50 values, lapatinib acted as a substrate for ABCB1 in both cases, thereby increasing its expression. On the contrary, based on the lower IC50 value of ABCG2, lapatinib acts as an inhibitor at the indicated dosages. Hypoxia-inducible factor 1α (HIF1α) is also known to induce the expression of ABCG2 [61], which may be attributed to the increased expression of ABCG2 observed in spheroids. Another possible mechanism is Twist1-mediated elevation of (ABC) transporters. Previously, Li et al. reported Twist1-mediated upregulation of EMT and MDR in breast cancer [62]. The mechanism for the here observed increased expression of ABCC1 by XAV939, however, remains to be resolved. We hypothesize that the contrasting result obtained for lapatinib on monolayers and spheroid models regarding EMT regulation may be due to dose differences, resulting in differential activities of ABC transporters. At a 6-fold higher dose of lapatinib in the spheroids, these transporters are inhibited, leading to decreased expression of EMT markers. Although MDM2 and caveolin-1 are known to be involved in multiple cellular processes, they are also implicated in instigating EMT and drug resistance. We found that lapatinib and XAV939 combination therapy reduced their accumulation both in monolayers and spheroids. Therefore, our findings indicate multiple connections between EMT and ABC transporters mediated by MDR.
Our results suggest that, in addition to other mechanisms that might be at play, inhibition of EMT contributes to inhibition of the migratory and invasive properties of mesenchymal-like TNBC cells. In line with this, Vijay et al. observed a decreased migration of TNBC cells after targeting EMT by inhibition of GSK-3β [63]. We therefore focused on selected elements of the EGFR and Wnt/β-catenin pathways as primary mediators of pro-survival/anti-apoptotic/pro-migration/drug resistance signaling. After incubation with lapatinib, XAV939 and their combination, the pEGFR level was found to be reduced significantly. The effect of the combination was found to be profound in spheroids. Reduction of the pEGFR level by XAV939 suggested Wnt-mediated transactivation of EGFR, probably by MMP-mediated release of soluble EGFR ligand [64]. The unaltered level of β-catenin after administration of XAV939 alone indicated that this inhibitor could only exert its profound effect at high concentrations. Consistent with the report by Tian et al., where XAV939 decreased the expression of β-catenin, we found that application of both inhibitors markedly downregulated β-catenin expression. This indicates a high responsiveness of the new therapeutic approach [55]. Concomitant reversal of EMT following decreases in pEGFR and β-catenin levels implies the importance of these pathways in TNBC aggressiveness.
Stimulation of EGFR activates pro-oncogenic MAPK, STAT-3 and AKT, which have been found to be highly dysregulated in metastatic TNBC [65,66,67]. The Wnt/β-catenin pathway shares a bidirectional positive feedback loop with MAPK, STAT3 and AKT, primarily through GSK3β [68,69,70]. Cell survival signals inactivate GSK3β through phosphorylation and, thereby, stabilize β-catenin [71]. Earlier studies have confirmed a role of MAPK, STAT-3 and AKT in EMT [10, 67, 68, 72, 73]. The observed decline in expression of pMAPK, pSTAT-3, pAKT and pGSK3β, vital downstream components of the EGFR and Wnt signaling pathways, after incubation with the inhibitors, signifies the complexity and web-like connectivity between the examined receptors and mark the inhibition of EMT and growth signals. These findings are clear indicators of synergistic anti-cancer cell survival actions of the combination treatment through downstream signaling.
5 Conclusions
Aberrant activation of EMT and associated cancer stem cell-like phenotypes are the major causes of resistance to therapy in TNBC. In the pursuit of effective therapy, we resorted to the co-treatment module using lapatinib and XAV939, which target the EGFR and Wnt/β-catenin pathways, two pivotal signaling pathways underlying EMT. Concurrent with reversal of the EMT phenotype by co-treatment, expression of genes responsible for cancer stemness and MDR were also curbed. Additionally, inhibition of EMT resulted in reduced migration and invasion of TNBC cells. Nonetheless, our proposed combination therapy was able to circumvent most of the complexities by targeting multiple signaling nodes and preventing downstream crosstalk. Overall, our results underscore a potential combined application of EGFR and Wnt/β-catenin inhibitors in the targeted therapy of TNBC.
References
J.M. Lee, S. Dedhar, R. Kalluri, E.W. Thompson, The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol. 172, 973–981 (2006)
V.L. Battula, K.W. Evans, B.G. Hollier, Y. Shi, F.C. Marini, A. Ayyanan, Ry. Wang, C. Brisken, R. Guerra, M. Andreeff, Epithelial-mesenchymal transition‐derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 28, 1435–1445 (2010)
S. Lamouille, J.X.R. Derynck, Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 15, 178–196 (2014)
J.P. Thiery, H. Acloque, R.Y. HuangM.A. Nieto, Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009)
G.V. Røsland, S.E. Dyrstad, D. Tusubira, R. Helwa, T.Z. Tan, M.L. Lotsberg, I.K. Pettersen, A. Berg, C. Kindt, F. Hoel, Epithelial to mesenchymal transition (EMT) is associated with attenuation of succinate dehydrogenase (SDH) in breast cancer through reduced expression of SDHC. Cancer Metab. 7, 6 (2019)
S.K. Singh, C. Hawkins, I.D. Clarke, J.A. Squire, J. Bayani, T. Hide, R.M. Henkelman, M.D. Cusimano, P.B. Dirks, Identification of human brain tumour initiating cells. Nature 432, 396 (2004)
A. Singh, J. Settleman, EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29, 4741 (2010)
R.-R. BegicevicM. Falasca, ABC transporters in cancer stem cells: Beyond chemoresistance. Int. J. Mol. Sci. 18, 2362 (2017)
G. Palma, G. Frasci, A. Chirico, E. Esposito, C. Siani, C. Saturnino, C. Arra, G. Ciliberto, A. GiordanoM, D’Aiuto, Triple negative breast cancer: looking for the missing link between biology and treatments. Oncotarget 6, 26560 (2015)
J.M. Dolle, J.R. Daling, E. White, L.A. Brinton, D.R. Doody, P.L. PorterK.E. Malone, Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer Epidem. Biomar. 18, 1157–1166 (2009)
J.S. Lee, S.E. Yost, S. Blanchard, D. Schmolze, H.H. Yin, R. Pillai, K. Robinson, A. Tang, N. Martinez, J. Portnow, Phase I clinical trial of the combination of eribulin and everolimus in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 21, 119 (2019)
H.S. Park, M.H. Jang, E.J. Kim, H.J. Kim, H.J. Lee, Y.J. Kim, J.H. Kim, E. Kang, S.-W. Kim, I.A. Kim, High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 27, 1212 (2014)
A. Simiczyjew, E. Dratkiewicz, M. Van Troys, C. Ampe, I. Styczeń, D. Nowak, Combination of EGFR inhibitor Lapatinib and MET inhibitor Foretinib inhibits migration of triple negative breast cancer cell lines. Cancers (Basel)10, 335 (2018)
A. Gangrade, V. Pathak, C. Augelli-Szafran, H.-X. Wei, P. Oliver, M.S.D. Buchsbaum, Preferential inhibition of Wnt/β-Catenin signaling by novel Benzimidazole compounds in triple-negative breast cancer. Int. J. Mol. Sci. 19, 1524 (2018)
T.H.C. Li, Convergence between Wnt-β-catenin and EGFR signaling in cancer. Mol. Cancer 9, 236 (2010)
S.-M.A. Huang, Y.M. Mishina, S. Liu, A. Cheung, F. Stegmeier, G.A. Michaud, O. Charlat, E. Wiellette, Y. Zhang, S. Wiessner, Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614 (2009)
A. Walzl, C. Unger, N. Kramer, D. Unterleuthner, M. Scherzer, M. Hengstschläger, D. Schwanzer-Pfeiffer, H. Dolznig, The resazurin reduction assay can distinguish cytotoxic from cytostatic compounds in spheroid screening assays. J. Biomol. Screen. 19, 1047–1059 (2014)
T.-C. Chou, P. Talalay, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22, 27–55 (1984)
C.R.I. Nicoletti, Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 1, 1458 (2006)
A.I.M. Kubbies, Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. J. Biomol. Screen. 11, 922–932 (2006)
J. Friedrich, C. Seidel, R. Ebner, L.A. Kunz-Schughart, Spheroid-based drug screen: considerations and practical approach. Nat. Protoc. 4, 309–324 (2009)
X. Rao, X. Huang, Z.Z.X. Lin, An improvement of the 2ˆ (–delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 3, 71 (2013)
H. Wu, S. Wang, D. Weng, H. Xing, X. Song, T. Zhu, X. Xia, Y. Weng, XuL. Meng, Reversal of the malignant phenotype of ovarian cancer A2780 cells through transfection with wild-type PTEN gene. Cancer Lett. 271, 205–214 (2008)
W.-L. Chen, K.-T. Kuo, T.-Y. Chou, C.-L. Chen, C.-H. Wang, Y.-H. Wei, L.-S. Wang, The role of cytochrome c oxidase subunit Va in non-small cell lung carcinoma cells: association with migration, invasion and prediction of distant metastasis. BMC Cancer 12, 273 (2012)
B. Bao, C. Mitrea, P. Wijesinghe, L. Marchetti, E. Girsch, R.L. Farr, J.L. Boerner, R. Mohammad, G. Dyson, S.R. Terlecky, Treating triple negative breast cancer cells with erlotinib plus a select antioxidant overcomes drug resistance by targeting cancer cell heterogeneity. Sci. Rep. 7, 44125 (2017)
A.S. Nunes, A.S. Barros, E.C. Costa, A.F. MoreiraI, J. Correia, 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 116, 206–226 (2019)
B.R. Topacio, E. Zatulovskiy, S. Cristea, S. Xie, C.S. Tambo, S.M. Rubin, J. Sage, M. Kõivomägi, J.M. Skotheim, Cyclin D-Cdk4,6 drives cell-cycle progression via the Retinoblastoma protein’s C-terminal helix. Mol. Cell 74, 758-770.e754 (2019)
M. Redza-Dutordoir, A.D. Averill-Bates, Activation of apoptosis signalling pathways by reactive oxygen species. BBA-Mol. Cell Res. 1863, 2977–2992 (2016)
J.M. Matés, F.M. Sánchez-Jiménez, Role of reactive oxygen species in apoptosis: implications for cancer therapy. Int. J. Biochem. Cell. Biol. 32, 157–170 (2000)
A. Perelman, C. Wachtel, M. Cohen, S. Haupt, H. Shapiro, A. Tzur, JC-1: alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 3, e430 (2012)
N. Pećina-Šlaus, Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 3, 17 (2003)
A.S.S. Li, Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 68, 3033–3046 (2011)
K.M. Mrozik, O.W. Blaschuk, C.M. Cheong, A.C.W. Zannettino, K. Vandyke, N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 18, 939 (2018)
M.-H. Wang, R. Sun, X.-M. Zhou, M.-Y. Zhang, J.-B. Lu, Y. Yang, L.-S. Zeng, X.-Z. Yang, L. Shi, R.-W. Xiao, Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 9, 2 (2018)
U.M. Moll, O. Petrenko, The MDM2-p53 interaction. Mol. Cancer Res. 1, 1001-1008 (2003)
W.S.L. Tang, MDM2 increases drug resistance in cancer cells by inducing EMT independent of p53. Curr. Med. Chem. 23, 4529–4539 (2016)
Z.C. Nwosu, M.P. Ebert, S.D.C. Meyer, Caveolin-1 in the regulation of cell metabolism: a cancer perspective. Mol. Cancer 15, 71 (2016)
C. Dai, A.K. Tiwari, C.-P. Wu, X. Su, S.-R. Wang, D. Liu, C.R. Ashby, Y. Huang, R.W. Robey , Y.-J. Liang, Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 68, 7905–7914 (2008)
D. Fang, D. Hawke, Y. Zheng, Y. Xia, J. Meisenhelder, H. Nika, G.B. Mills, R. Kobayashi, T. Hunter, Z. Lu, Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 (2007)
C. Garay, G. Judge, S. Lucarelli, S. Bautista, R. Pandey, T. Singh, C.N. Antonescu, Epidermal growth factor-stimulated Akt phosphorylation requires clathrin or ErbB2 but not receptor endocytosis. Mol. Biol. Cell 26, 3504–3519 (2015)
Y. Wang, S. Pennock, X.C.Z. Wang, Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol. Cell. Biol. 22, 7279 (2002)
Q. Ryan, A. Ibrahim, M.H. Cohen, J. Johnson, C. Ko, R. Sridhara, R. Justice, R. Pazdur, FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 13, 1114–1119 (2008)
N.K.R. Kurzrock, Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 62, 50–60 (2018)
M. Kahn, Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 (2014)
A.I. MinchintonI.F. Tannock, Drug penetration in solid tumours. Nat. Rev. Cancer 6, 583–592 (2006)
E.C. Costa, A.F. Moreira, D. de Melo-Diogo, V.M. Gaspar, M.P. Carvalho, I.J. Correia, 3D tumor spheroids: an overview on the tools and techniques used for their analysis. Biotechnol. Adv. 34, 1427–1441 (2016)
O. Tetsu, F. McCormick, β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 (1999)
I. Hadžisejdić, E. Mustać, N. Jonjić, M. Petković, B. Grahovac, Nuclear EGFR in ductal invasive breast cancer: correlation with cyclin-D1 and prognosis. Mod. Pathol. 23, 392–403 (2010)
C.P. Masamha, D.M. Benbrook, Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of Cyclin E2 in ovarian cancer cells. Cancer Res. 69, 6565 (2009)
Y. Jiang, X.W.D. Hu, Furanodienone induces G0/G1 arrest and causes apoptosis via the ROS/MAPKs-mediated caspase-dependent pathway in human colorectal cancer cells: a study in vitro and in vivo. Cell Death Dis. 8, e2815–e2815 (2017)
H.-R. Teppo, Y. Soini, P. Karihtala, Reactive oxygen species-mediated mechanisms of action of targeted cancer therapy. Oxid. Med. Cell. Longev. 2017, 1485283 (2017)
K.M. Aird, J.L. Allensworth, I. Batinic-Haberle, H.K. Lyerly, M.W. Dewhirst, G.R. Devi, ErbB1/2 tyrosine kinase inhibitor mediates oxidative stress-induced apoptosis in inflammatory breast cancer cells. Breast Cancer Res. Treat. 132, 109–119 (2012)
J. Chandra, A.S.S. Orrenius, Triggering and modulation of apoptosis by oxidative stress. Free Radical Biol. Med. 29, 323–333 (2000)
R. Nahta, L.X. Yuan, Y. Du, F.J. Esteva, Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol. Cancer Ther. 6, 667–674 (2007)
X.-H. Tian, W.-J. Hou, Y. Fang, J. Fan, H. Tong, S.-L. Bai, Q. Chen, H. Xu, Y. Li, XAV939, a tankyrase 1 inhibitior, promotes cell apoptosis in neuroblastoma cell lines by inhibiting Wnt/β-catenin signaling pathway. J. Exp. Clin. Cancer Res. 32, 100 (2013)
C.-Y. Liu, M.-H. Hu, C.-J. Hsu, C.-T. Huang, D.-S. Wang, W.-C. Tsai, Y.-T. Chen, C.-H. Lee, P.-Y. Chu, C.-C. Hsu, Lapatinib inhibits CIP2A/PP2A/p-Akt signaling and induces apoptosis in triple negative breast cancer cells. Oncotarget 7, 9135 (2016)
K.B. Yin, The Mesenchymal-Like Phenotype of the MDA-MB-231 Cell Line. In: M. Gunduz, E. Gunduz ed. Breast Cancer - Focusing Tumor Microenvironment, Stem cells and Metastasis. IntechOpen 385–402 (2011). https://doi.org/10.5772/20666. Available from:https://www.intechopen.com/books/breast-cancer-focusing-tumor-microenvironment-stem-cells-and-metastasis/the-mesenchymal-like-phenotype-of-themda-mb-231-cell-line
Y.-C. Hsiao, M.-H. Yeh, Y.-J. Chen, J.-F. Liu, C.-H. Tang, W.-C. Huang, Lapatinib increases motility of triple-negative breast cancer cells by decreasing miRNA-7 and inducing Raf-1/MAPK-dependent interleukin-6. Oncotarget 6, 37965 (2015)
Z.C. Hartman, G.M. Poage, P. Den Hollander, A. Tsimelzon, J. Hill, N. Panupinthu, Y. Zhang, A. Mazumdar, S.G. Hilsenbeck, G.B. Mills, Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 73, 3470–3480 (2013)
J.W. Polli, J.E. Humphreys, K.A. Harmon, S. Castellino, M.J. O’mara, K.L. Olson, L.S. John-Williams, K.M. Koch, C.J. Serabjit-Singh, The role of efflux and uptake transporters in N-{3-chloro-4-[(3-fluorobenzyl) oxy] phenyl}-6-[5-({[2-(methylsulfonyl) ethyl] amino} methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab. Disposition 36, 695–701 (2008)
X. He, J. Wang, W. Wei, M. Shi, B. Xin, T.Z.X. Shen, Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1α and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 17, 188–198 (2016)
Q.-Q. Li, J.-D. Xu, W.-J. Wang, X.-X. Cao, Q. Chen, F. Tang, Z.-Q. Chen, X.-P. Liu, Z.-D. Xu, Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer. Res. 15, 2657–2665 (2009)
G.V. Vijay, N. Zhao, P. Den Hollander, M.J. Toneff, R. Joseph, M. Pietila, J.H. Taube, T.R. Sarkar, E. Ramirez-Pena, S.J. Werden, GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 21, 37 (2019)
G. Civenni, T. Holbro, N.E. Hynes, Wnt1 and Wnt5a induce cyclin D1 expression through ErbB1 transactivation in HC11 mammary epithelial cells. EMBO Rep. 4, 166–171 (2003)
P.W.Z. Wang, Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel)9, 52 (2017)
M.K. Ghosh, P. Sharma, P.C. Harbor, S.O. RahamanS.J. Haque, PI3K-AKT pathway negatively controls EGFR-dependent DNA-binding activity of Stat3 in glioblastoma multiforme cells. Oncogene 24, 7290 (2005)
J. Gyamfi, Y.-H. Lee, M. Eom, J. Choi, Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 8, 8859 (2018)
W.-J. Jeong, E.J. Ro, K.-Y. Choi, Interaction between Wnt/β-catenin and RAS-ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. NPJ Precis. Oncol. 2, 1–10 (2018)
M.A. Fragoso, A.K. Patel, R.E. Nakamura, H. Yi, K. Surapaneni, A.S. Hackam, The Wnt/β-catenin pathway cross-talks with STAT3 signaling to regulate survival of retinal pigment epithelium cells. PLoS One 7, e46892 (2012)
N.T. Georgopoulos, L.A. Kirkwood, J. Southgate, A novel bidirectional positive-feedback loop between Wnt–β-catenin and EGFR–ERK plays a role in context-specific modulation of epithelial tissue regeneration. J. Cell Sci. 127, 2967–2982 (2014)
C. Metcalfe, C. Mendoza-Topaz, J.M.M. Bienz, Stability elements in the LRP6 cytoplasmic tail confer efficient signalling upon DIX-dependent polymerization. J. Cell Sci. 123, 1588–1599 (2010)
S.J. Grille, A. Bellacosa, J. Upson, A.J. Klein-Szanto, F. Van Roy, W. Lee-Kwon, M. Donowitz, P.N. Tsichlis, L. Larue, The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 63, 2172–2178 (2003)
H. Zheng, W. Li, Y. Wang, Z. Liu, Y. Cai, T. Xie, M. Shi, Z. Wang, B. Jiang, Glycogen synthase kinase-3 beta regulates Snail and β-catenin expression during Fas-induced epithelial–mesenchymal transition in gastrointestinal cancer. Eur. J. Cancer 49, 2734–2746 (2013)
Acknowledgements
The authors acknowledge the DBT program Support Facility, Centre for Nanotechnology, Department of Electronics and Information Technology grant, and the Central Instruments Facility (CIF), IIT Guwahati, India. The authors also acknowledge Ms. Satakshi Hazra for her help in editing the manuscript.
Funding
The study was supported by grants from the Department of Biotechnology, Government of India (BT/PR13560/COE/34/44/2015, and BT/PR 25095/NER/95/1011/2017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they do not have any conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 4.02 MB)
Rights and permissions
About this article

Cite this article
Shome, R., Ghosh, S.S. Tweaking EMT and MDR dynamics to constrain triple-negative breast cancer invasiveness by EGFR and Wnt/β-catenin signaling regulation. Cell Oncol. 44, 405–422 (2021). https://doi.org/10.1007/s13402-020-00576-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-020-00576-8