
Abstract
Background
MicroRNAs (miRNAs) are known to play an important role in cancer development by post-transcriptionally affecting the expression of critical genes. The aims of this study were two-fold: (i) to develop a robust method to isolate miRNAs from small volumes of saliva and (ii) to develop a panel of saliva-based diagnostic biomarkers for the detection of head and neck squamous cell carcinoma (HNSCC).
Methods
Five differentially expressed miRNAs were selected from miScript™ miRNA microarray data generated using saliva from five HNSCC patients and five healthy controls. Their differential expression was subsequently confirmed by RT-qPCR using saliva samples from healthy controls (n = 56) and HNSCC patients (n = 56). These samples were divided into two different cohorts, i.e., a first confirmatory cohort (n = 21) and a second independent validation cohort (n = 35), to narrow down the miRNA diagnostic panel to three miRNAs: miR-9, miR-134 and miR-191. This diagnostic panel was independently validated using HNSCC miRNA expression data from The Cancer Genome Atlas (TCGA), encompassing 334 tumours and 39 adjacent normal tissues. Receiver operating characteristic (ROC) curve analysis was performed to assess the diagnostic capacity of the panel.
Results
On average 60 ng/μL miRNA was isolated from 200 μL of saliva. Overall a good correlation was observed between the microarray data and the RT-qPCR data. We found that miR-9 (P <0.0001), miR-134 (P <0.0001) and miR-191 (P <0.001) were differentially expressed between saliva from HNSCC patients and healthy controls, and that these miRNAs provided a good discriminative capacity with area under the curve (AUC) values of 0.85 (P <0.0001), 0.74 (P < 0.001) and 0.98 (P < 0.0001), respectively. In addition, we found that the salivary miRNA data showed a good correlation with the TCGA miRNA data, thereby providing an independent validation.
Conclusions
We show that we have developed a reliable method to isolate miRNAs from small volumes of saliva, and that the saliva-derived miRNAs miR-9, miR-134 and miR-191 may serve as novel biomarkers to reliably detect HNSCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in the world [1–3] and annually approximately 900,000 new cases are reported worldwide, including 300,000 deaths [4]. HNSCC is diverse in nature, both biologically and clinically. The primary risk factors for the development of HNSCC include tobacco use, alcohol consumption, human papilloma virus (HPV) infection (for oropharyngeal cancer) and Epstein-Barr virus (EBV) infection (for nasopharyngeal cancer). In the initial stages of the disease a patient may not show any clinical symptoms and, consequently, a significant number of patients present with metastatic disease at the time of diagnosis (regional nodal involvement in 43 % and distant metastasis in 10 %), leading to 5-years survival rates of less than 40 % [5]. The early diagnosis of HNSCC holds promise for an improved prognosis, but this option is currently impeded by the lack of clinically validated biomarkers to reliably detect the disease at an early stage and the paucity of targeted molecular therapeutics [6].
Recently, the use of saliva to detect different pathologies, including cancer, has been shown to hold great promise [6–10]. Indeed, saliva sampling is rapidly expanding and may provide a non-invasive and cheap alternative to traditional tissue and blood sampling [11]. The ease to conduct saliva-based tests also turns it into an attractive option for large scale population-based screening and repeated sampling to monitor and manage disease progression [12]. Li et al. [13] reported that the salivary transcriptome contains more than 3,000 RNA species, mostly mRNAs, and Park et al. [14] and Patel et al. [15] reported the possibility of using salivary microRNAs (miRNAs) to detect oral cancers. Additionally, Wong et al. [16] emphasized the potential use of these miRNAs as biomarkers for cancer screening programs.
miRNAs are small noncoding RNAs (approximately 22 nucleotides long) that are known to play a crucial role in the post-transcriptional regulation of gene expression by either inhibiting target mRNA translation and/or by degrading mRNA targets [17, 18]. To date, the miRNA registry contains 1,872 human miRNA precursors, giving rise to 2,578 mature miRNAs (MiRBase, Release 20), which are thought to regulate at least 30 % of all human genes. Within the human genome miRNAs constitute 3-5 % of the predicted genes, and they can be located in intergenic, intronic or exonic regions of either protein-coding or non-protein-coding genes [19]. miRNAs are frequently located in cancer-associated genomic regions [20] and have been shown to govern important cellular process such as proliferation, differentiation, apoptosis and cell cycle regulation. As such, miRNAs may play a crucial role in cancer initiation, progression and/or metastasis [21].
From an initial study, using miScript™ miRNA PCR Arrays from Qiagen, we selected a subset of five differentially expressed miRNAs (based on > 2-fold change) to determine their clinical utility. Our aims were (i) to develop a robust method to isolate miRNAs from small volumes of saliva and (ii) to develop a novel salivary biomarker panel for the early detection of HNSCC.
2 Materials and methods
2.1 Participants and samples
Unstimulated saliva samples were collected from HNSCC patients (n = 61, including the 5 samples used in the miRNA PCR arrays) at various stages (II-IV) of the disease, who presented to the Head and Neck Clinic at the Princess Alexandra Hospital, Woolloongabba, Australia. Similarly, saliva samples were collected from healthy individuals (n = 61, including the 5 samples used in the miRNA PCR arrays) with no previous history of any malignancy, and who were non-smokers with a good oral hygiene. All samples were immediately stored in dry ice and transferred to the laboratory for processing. The participants’ demographics are listed in Table 1 and their clinical data are listed in supplementary Table 1. All participants provided written informed consent before sample collection. This study was approved by the University of Queensland Medical Ethical Institutional Board and by the Princess Alexandra Hospital Ethics Review Board.
2.2 Sample characteristics
We collected whole mouth resting saliva in sterile 50 mL Falcon tubes (Sarstedt, Australia) as reported before [7, 22]. Cell-free salivary supernatants allow the detection of miRNAs that are secreted into the saliva, whereas whole mouth salivas allow the detection of both secretorty and non-secretory miRNAs. Park et al. [14] concluded from their work that the miRNA levels in salivary cell-free supernatants are lower than those in whole mouth salivas and, in addition, that more heterogeneous miRNA populations can be found in whole saliva. Moreover, Patel et al. [15] reported that they were able to obtain high resolution microRNA signatures from whole saliva samples.
2.3 miRNA extraction and cDNA synthesis
QIAzol lysis reagent (Qiagen, Valencia, CA, USA) was used as an extraction solvent to isolate total RNA and a NucleoSpin miRNA kit (Macherey-Nagel, Düren, Germany) was used to enrich for miRNAs. When using a NucleoSpin miRNA kit, there are 3 options: (i) to isolate total RNA (miRNAs and large RNAs) in the same fraction, (ii) to isolate miRNAs and large RNAs in two separate fractions or (iii) to only isolate miRNAs. We have used option (iii). In brief, 800 μL QIAzol lysis reagent was added to 200 μL whole saliva. This mixture was briefly vortexed and incubated for 5 min at room temperature (RT). Next, 200 μL chloroform was added, and the mixture was vortexed vigorously and incubated at RT for another 5 min. The samples were then centrifuged at 10,000 × g for 10 min at 4 °C. The upper aqueous layer (800 μL) was transferred into a new microcentrifuge tube, another 200 μL chloroform was added and the mixture was vortexed again. After this, the samples were again centrifuged at 10,000 × g at 4 °C. The upper aqueous layer (800 μL), containing total RNA, was carefully removed and transferred to a new microcentrifuge tube and 200 μL 100 % ethanol was added. This mixture was used for miRNA enrichment using a NucleoSpin miRNA kit. To this end, the solution containing total RNA was passed through a Nucleospin column (blue ring) by centrifugation at 11,000 × g for 30 s at RT. The flow through was collected and 800 μL MX buffer was added. Next, the sample was loaded onto a Nucleospin column (green ring) and centrifuged at 11,000 × g for 30 s at RT. The columns were then washed with 600 μL MW1 buffer and centrifuged at 11,000 × g for 30 s. The wash was repeated with 700 μL MW1 buffer followed by 250 μL MW2 buffer and centrifugation at 11,000 × g for 30 s. The columns were centrifuged at 11,000 × g for another 2 min to remove all the ethanol. Then, the miRNA was eluted in 30 μL RNase free water. The quantity of the isolated miRNA was determined using a Nanodrop ND-1,000 spectrophotometer (Thermo scientific Wilmington DE). In supplementary Table 2 the concentrations of the miRNAs are listed. OD 260:280 ratios ≥ 1.8 were accepted as pure.
Using the isolated miRNAs as template (input 250 ng), cDNA synthesis was carried out via a miScript II RT Kit with Hispec buffer (Qiagen, Valencia, CA, USA) according to the manufacturers’ instructions. In brief, miRNAs were polyadenylated with polymerase A and transcribed into cDNA using oligo-dT primers (in parallel in the same tube). The oligo-dT had a 3′degenerate anchor and a universal tag sequence at the 5′end allowing amplification of mature miRNA through a real-time PCR step. The polyadenylation and universal tag primer ensure that genomic DNA is not amplified. Therefore, DNAase treatment was omitted.
2.4 miRNA PCR array assay
miScript™ miRNA PCR arrays (Qiagen, catalogue number MIHS-3102Z) were used according to the manufactur’s instructions. Briefly, 100 μl saliva samples from five HNSCC patients and from five healthy controls were pooled separately to reduce biological variation. From these two saliva sample pools, miRNA was extracted and converted to cDNA as described above. Next, the cDNA was diluted 1:10 in RNAse/DNAse free water and used as a template for miRNA PCR array amplification. Before that, the PCR array plate was sealed and centrifuged at 1,000 × g for 1 min at RT to remove air bubbles that may interfere with the PCR amplification process. Then, PCR reactions were performed using a thermo cycler (Bio-Rad, Hercules, CA, USA). The reaction mixtures were incubated at 95 °C for 10 min to activate the HotStart DNA Taq polymerase, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s, monitored by melt curve analysis. Ct values >35 were excluded from the analyses.
miScript™ miRNA PCR Arrays allow the profiling of 84 miRNAs that were selected based their presumed correlation with the diagnosis, staging, progression and/or prognosis of various cancers. A set of controls present on this array enables data analysis using the ΔΔCt method, the assessment of reverse transcription performance and the assessment of PCR performance. The endogenous normaliser SNORD96A [23, 24] showed a relatively stable expression across control and HNSCC patient samples. The miScript miRNA PCR Array Data Analysis Web Portal (http://pcrdataanalysis.sabiosciences.com/mirna/arrayanalysis.php) was used to analyse the microarray data (Qiagen, Germany).
2.5 Quantitative real-time PCR (RT-qPCR)
To confirm the microarray-based expression data, saliva collected from 21 HNSCC patients and 21 healthy controls were first analysed using RT-qPCR. Next, we expanded the cohorts of HNSCC patients and controls with 35 each to validate the clinical utility of the identified miRNAs. To this end, the cDNAs were diluted 1:10 in RNAse/DNAse free water and used as a template for PCR amplification. The PCRs were run in duplicate using a miScript Syber green PCR master mix (Qiagen, Valencia, CA, USA) and carried out in a thermo cycler (Bio-Rad, Hercules, CA, USA) The reaction mixtures were incubated at 95 °C for 15 min to activate the hot start Taq DNA polymerase, followed by 40 cycles of 94 °C for 15 s, 55 °C for 30s and 70 °C for 30s, monitored by melt curve analysis. According to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines [25], the threshold value (Ct) was determined for each well and the values were averaged for each gene (see supplementary Table 3). Samples with Ct values > 35 were excluded from the analysis.
2.6 Independent validation using TCGA data
Additionally, The Cancer Genome Atlas (TCGA) miRNAseq data were used to validate our findings. In our study, we used saliva samples for miRNA profiling. There are two ways by which tumour-specific molecules can enter the saliva (i) via direct release or (ii) via the secretion of exsosomes/microvesicles [26–29]. From the TCGA data portal (HNSCC cancer) we downloaded Illumina HiSeq miRNAseq data from 334 tumours and 39 normal tissues, and performed PCA expression analysis (see below) for both tumour and normal tissues. We excluded 8 samples (4 from tumours and 4 from normal tissues) that were found to be outliers.
2.7 Statistical analysis
All statistical analyses were performed using GraphPad Prism 5 software version 5.03 (GraphPad Software Inc., USA). Data analyses were performed using the miScript miRNA PCR Array Data Analysis Excel web portal (http://pcrdataanalysis.sabiosciences.com/mirna/arrayanalysis.php), including quantification by the ΔΔCT method [30]. Data normalization was performed using an endogenous control (SNORD96A) to correct for sample variation. miRNA expression levels were compared using the Mann Whitney U- test for non-parametric analysis. Receiver-operating characteristics (ROC) curves were generated for five selected miRNAs to determine their clinical utility as diagnostic biomarkers. For TCGA data analysis, differential expression was evaluated on the reads per million counts (on a log2 scale) using the Wilcoxon rank sum test. The Bonferroni method was used for multiple hypothesis correction.
3 Results
3.1 High miRNA yields from small saliva samples
We have established a robust and reproducible method to isolate high yield miRNAs from both fresh and archival saliva samples based on QIAzol (Qiagen) extraction followed by solid phase enrichment of miRNAs on silica columns. This method allowed us to isolate miRNAs from as little as 200 μL of whole mouth saliva. This method also turned out to be robust for isolating high yield miRNAs from archival saliva samples stored at −80 °C for up to 2 years. Supplementary Table 2 depicts the concentrations and the OD 260/280 measurements for the individual samples. The amounts of miRNAs isolated from 200 μL of saliva ranged from 11.50 ng to 644 ng.
3.2 miRNA expression in saliva from HNSCC patients and healthy controls
After miScript™ miRNA PCR Array based expression analysis using pooled samples from five HNSCC patients and five healthy controls, a subset of five differentially expressed miRNAs (based on > 2-fold change) was selected for confirmation and validation in independent (the first 5 pooled samples are included in these cohorts: see material and methods and supplementary Tables 1, 2, and 3) cohorts using RT-qPCR. These miRNAs were: miR-9, miR-127, miR-134, miR-191 and miR-222. For the confirmatory study we used saliva from 21 HNSCC patients and 21 healthy controls and for the validation study we used saliva from an independent cohort of 35 HNSCC patients and 35 healthy controls. When comparing the microarray and RT-qPCR results side-by-side after normalizing both data sets with the same control (SNORD96A) we found that, with the exception of miR-191, in the confirmatory cohort (n = 21) all other miRNA microarray data were in agreement with the RT-qPCR data (Fig. 1a). The average SNORD96A Ct values for the saliva collected from the healthy controls (Ct = 22.9) and the HNSCC patients (Ct = 22.5) were similar (Mann–Whitney U Test), thus confirming its applicability as a normaliser (see supplementary Table 3). In Fig. 1b the RT-qPCR based fold-changes for the five selected miRNAs are listed. miR-127 (P = 0.07), miR-222 (P = 0.11), miR-191 (P < 0.01) and miR-9 (P = 0.68) were found to be up-regulated in the saliva from HNSCC patients as compared to the healthy controls (fold changes 2.3, 1.92, 8.2 and 3.7, respectively). In contrast, miR-134 was found to be down-regulated in the microarray data set but not in the RT-qPCR data set. In the second independent validation study (n = 35), except for miR-134, the other four miRNAs were consistently up-regulated in saliva from the HNSCC patients as compared to the saliva from the healthy controls (Fig. 2a).

a Relative expression levels of five miRNAs determined by microarray and RT-qPCR technologies, respectively. Microarray and RT-qPCR data sets were normalized to SNORD96A. b Expression levels of miR-222, miR-134, miR-127, miR-9 and miR-191 in saliva collected from HNSCC patients (n = 21) and healthy controls (n = 21) determined by RT-qPCR (confirmatory study)

a Expression levels of miR-222, miR-134, miR-127, miR-9 and miR-191 in saliva collected from HNSCC patients (n = 35) and healthy controls (n = 35) determined by RT-qPCR. Differences were determined using Wilcoxon Statistical test and statistical significance was obtained for miR-191, miR-9 and miR-134 (P < 0.001). b Illumina HiSeq miRNAseq data were downloaded from The Cancer Genome Atlas (TCGA) portal (HNSCC) of 334 tumours and 39 normal tissues. The Y-axis represents reads per million and the X-axis represents five selected miRNAs investigated
3.3 Independent validation using TCGA data
TCGA HNSCC miRNAseq data revealed that four of the five miRNAs: (miR-9, miR-127, miR-191 and miR-222) were differentially expressed (P < 0.01) after multiple hypothesis correction. miR-9, miR-191 and miRNA-222 showed a significant over-expression in tumours derived from HNSCC patients compared to normal tissues collected from the same patients. In contrast to salivary miRNA expression data for miRNA 127 and miRNA 134, both of these miRNAs were higher in normal tissues as compared to tumour samples from the same patients (Fig. 2b).
3.4 Discriminatory power of miR-9, miR-134 and miR-191
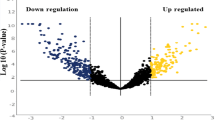
Receiver Operator Curves (ROC) were generated to evaluate the discriminatory power of miR-9, miR-134 and miR-191 for their ability to differentiate between control and disease groups. All three miRNAs were found to provide a good discriminative ability with AUC values of 0.85 (P < 0.0001), 0.74 (P < 0.001) and 0.98 (P < 0.0001), respectively. When combining all three miRNAs, the AUC was 0.74 (P < 0.0001). In contrast, miR-127 and miR-222 yielded AUCs values of 0.73 (P < 0.01) and 0.58 (P = 0.24), respectively. Figure 3 shows ROC analyses for these three miRNAs using salivary samples from all 56 HNSCC patients and healthy controls. Figure 4 depicts the signature cluster profiles of five selected salivary miRNAs.

Receiver operator characteristics (ROC) curve analysis of the independent validation study using saliva-derived miR-9 (a), miR-134 (b) and miR-191 (c) for discriminating HNSCC patients from normal subjects (P < 0.001)

miScriptTM microarray signature cluster profiles of five selected miRNAs in saliva from healthy controls (n = 56) and HNSCC patients (n = 56)
We also evaluated whether the miRNA expression profiles can discriminate HNSCC patients based on anatomical sites (Supplementary Table 4). To this end, again ROCs were generated. We found that the fold changes of miR-222 (P < 0.05), miR-127 (P < 0.05) and miR-191 (P < 0.05) could differentiate HNSCC patients belonging to Group 1 and Group 4 (Group 1 = oral cavity and Group 4 = pharynx). The expression of miR-9 could (P < 0.05) differentiate HNSCC patients in Group 2 and Group 3 (Group 2 = oropharynx and Group 3 = larynx) with an AUC of 0.77. Similarly, we found that the expression of miR-191 could (P < 0.05) differentiate HNSCC patients from Group 2 and Group 4 with an AUC of 0.94, and that the expression of miR-127 could (P < 0.01) differentiate HNSCC patients from Group 3 and Group 4 with an AUC of 1.00.
4 Discussion
Currently, the early diagnosis of head and neck squamous cell carcinoma (HNSCC) is severely hampered by the lack of suitable biomarkers. We have developed a protocol to isolate high yields of miRNAs from as little as 200 μL of fresh saliva, as well as from archival samples stored at −80 °C. By using these samples, we have identified and validated a novel salivary biomarker panel, consisting of miR-9, miR-191 and miR-134, to reliably detect HNSCC. Through an independent validation using the TCGA data base, the clinical utility of this panel was further confirmed.
Previously, Zhu et al. [31] reported that miR-9 over-expression may be associated with colorectal carcinoma metastasis by promoting cell motility. It was also noted that, when miR-9 was over-expressed in non-metastasising breast carcinoma cells, it was able to promote lung micro-metastases in a mouse model. Conversely, it was found that down-regulation of miR-9 in highly malignant cells may impair metastatic changes [32]. Furthermore, Hui et al. [33] reported that miR-9 and miR-9* were strongly associated with HPV/p16-status in oropharyngeal carcinomas. This observation is compatible with a report of Kozaki et al. [34] showing a significant up-regulation of miR-9 and miR-9* in oral squamous cell carcinoma (OSCC) cell lines. Khew-Goodall et al. [35] identified miR-9 as a negative regulator of E-cadherin by suppressing the ability to promote carcinoma cell motility and invasiveness.
We observed a down-regulation of miR-134 in the saliva samples from HNSCC patients. In contrast, Lin et al. [36] found that this miRNA was up-regulated in oral cancerous tissues compared to non-cancerous tissues, and that this up-regulation was associated with vascular invasion and tumour size. More recent studies have shown that miR-134 is involved in tumour suppression in prostate and lung cancers, and that its down-regulation is related to drug resistance [37].
Work carried out by Elyakim et al. [38], using a mouse model for hepatocellular carcinoma (HCC), has shown that dioxin (a liver carcinogen) can up-regulate miR-191 expression. Subsequent in vitro inhibition of miR-191 revealed a reduction in cellular proliferation and an increase in apoptosis. Similarly, a significant in vivo reduction in tumour load was observed. In addition, miR-191 was found to exhibit a tissue-specific expression pattern, and to be over-expressed in colon, lung, pancreas, prostate and stomach cancers [39]. Under hypoxic conditions, HNSCC cells showed an up-regulation of miR-191 [40]. Barker et al. [41] sought to establish a method to predict the origin of an unknown primary HNSCC tumour by utilising the expression of miR-191. In their study, miR-191 expression levels were found to be consistent between primary and local metastatic lesions within an individual patient and between different patients within a specific site [41]. They noted an association between primary and regional metastatic nodal sites in base-of-tongue and tonsillar carcinoma patients.
From our preliminary work we conclude that miR-9, miR-191 and miR-134 may serve as novel non-invasive biomarkers in HNSCC. Notably, the expression of salivary miR-9, miR-191 and miR-134 was independently validated using a HNSCC cohort consisting of 334 tumour samples and 39 normal adjacent tissues, curated in the TCGA database.
References
D. Chin, G.M. Boyle, S. Porceddu, D.R. Theile, P.G. Parsons, W.B. Coman, Head and neck cancer: past, present and future. Expert. Rev. Anticancer. Ther. 6, 1111–1118 (2006)
D. Weiss, C. Stockmann, K. Schrodter, C. Rudack, Protein expression and promoter methylation of the candidate biomarker TCF21 in head and neck squamous cell carcinoma. Cell. Oncol. 36, 213–224 (2013)
T. Nakaoka, A. Ota, T. Ono, S. Karnan, H. Konishi, A. Furuhashi, Y. Ohmura, Y. Yamada, Y. Hosokawa, Y. Kazaoka, Combined arsenic trioxide-cisplatin treatment enhances apoptosis in oral squamous cell carcinoma cells. Cell. Oncol. 37, 119–129 (2014)
P.A. Wingo, T. Tong, S. Bolden, Cancer statistics for African Americans. CA Cancer J. Clin. 45, 8–30 (1995)
D.G. Haller, L.D. Wagma, K.A. Camphausen and W.J. Hoskins, Cancer Management Handbook, 12th Edition. (Cancer Network, 2013), http://www.cancernetwork.com/cancer-management-12
T. Pfaffe, J. Cooper-White, P. Beyerlein, K. Kostner, C. Punyadeera, Diagnostic potential of saliva: current state and future applications. Clin. Chem. 57, 675–687 (2011)
R. Nagadia, P. Pandit, W.B. Coman, J. Cooper-White, C. Punyadeera, miRNAs in head and neck cancer revisited. Cell. Oncol. 36, 1–7 (2013)
U.M. Bailey, C. Punyadeera, J.J. Cooper-White, B.L. Schulz, Analysis of the extreme diversity of salivary alpha-amylase isoforms generated by physiological proteolysis using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 911, 21–26 (2012)
B.L. Schulz, J. Cooper-White, C.K. Punyadeera, Saliva proteome research: current status and future outlook. Crit. Rev. Biotechnol. 33, 246–59 (2012)
C. Punyadeera, P.D. Slowey, in Nanobiomaterials in clinical dentistry, ed. by K. Subramani, W. Ahmed, J.K. Hartsfield (William Andrew Publishing, Waltham, 2013), pp. 453–473
R.J. Genco, Salivary diagnostic tests. J. Am. Dent. Assoc. 143, 3S–5S (2012)
H. Fabryova, P. Celec, On the origin and diagnostic use of salivary RNA. Oral Dis. 20, 146–52 (2013)
Y. Li, X. Zhou, M.A. St John, D.T. Wong, RNA profiling of cell-free saliva using microarray technology. J. Dent. Res. 83, 199–203 (2004)
N.J. Park, H. Zhou, D. Elashoff, B.S. Henson, D.A. Kastratovic, E. Abemayor, D.T. Wong, Salivary microRNA: discovery, characterization, and clinical utility for oral cancer detection. Clin. Cancer Res. 15, 5473–5477 (2009)
R.S. Patel, A. Jakymiw, B. Yao, B.A. Pauley, W.C. Carcamo, J. Katz, J.Q. Cheng, E.K. Chan, High resolution of microRNA signatures in human whole saliva. Arch. Oral Biol. 56, 1506–1513 (2011)
D.T. Wong, Salivaomics. J. Am. Dent. Assoc. 143(10 Suppl), 19S–24S (2012)
S. Babashah, M. Sadeghizadeh, M.R. Tavirani, S. Farivar, M. Soleimani, Aberrant microRNA expression and its implications in the pathogenesis of leukemias. Cell. Oncol. 35, 317–334 (2012)
E. Yiannakopoulou, Targeting epigentic mechanisms and microRNAs by aspirin and other non steroidal anti-inflammatory agents-implications for cancer treatment and chemoprevention. Cell. Oncol. 37, 167–178 (2014)
D.P. Bartel, MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004)
G.A. Calin, C.D. Dumitru, M. Shimizu, R. Bichi, S. Zupo, E. Noch, H. Aldler, S. Rattan, M. Keating, K. Rai, L. Rassenti, T. Kipps, M. Negrini, F. Bullrich, C.M. Croce, Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lympocytic leukemmia. Proc. Natl. Acad. Sci. U. S. A. 99, 15524–15529 (2002)
D. Xiao, J. Ohlendorf, Y. Chen, D.D. Taylor, S.N. Rai, S. Waigel, W. Zacharias, H. Hao, K.M. McMasters, Identifying mRNA, microRNA and protein profiles of melanoma exosomes. PLoS One 7, e46874 (2012)
R. Mohamed, J.L. Campbell, J. Cooper-White, G. Dimeski, C. Punyadeera, The impact of saliva collection and processing methods on CRP, IgE, and Myoglobin immunoassays. Clin. Transl. Med. 1, 19 (2012)
Y. Choi, H.P. Dienes, K. Krawczynski, Kinetics of miR-122 expression in the liver during acute HCV infection. PLoS One 8, e76501 (2013)
S.C. Sahu, microRNAs in: Toxicology and Medicine, 1st edn. (Wiley, 2013), pg 486
S.A. Bustin, G.L. Shipley, J. Vandesompele, C.T. Wittwer, V. Benes, J.A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M.W. Pfaffl, The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009)
A.L. Carvalho, W.-W. Jiang, Q. Claybourne, Y. Tokumaru, J. Lee, D. Goldenberg, E. Garrett-Mayer, S. Goodman, C.-s. Moon, W. Koch, W.H. Westra, C. Jeronimo, D. Sidransky, J.A. Califano, M.M. Kim, R. Henrique, Z. Zhang, M.O. Hoque, S. Chang, M. Brait, C.S. Nayak, Evaluation of promoter hypermethylation detection in body fluid as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin. Cancer Res. 14, 97–107 (2008)
A.K. El-Naggar, K. Hurr, J.G. Batsakis, M.A. Luna, H. Goepfert, V. Huff, Sequential loss of heterozygosity at microsatellite motifs in preinvasive and invasive head and neck squamous carcinoma. Cancer Res. 55, 2656–2659 (1995)
M.F. Huang, Y.C. Chang, P.S. Liao, T.H. Huang, C.H. Tsay, M.Y. Chou, Loss of heterozygosity of p53 gene of oral cancer detected by exfoliative cytology. Oral Oncol. 35, 296–301 (1999)
A.K. El-Naggar, L. Mao, G. Staerkel, M.M. Coombes, S.L. Tucker, M.A. Luna, G.L. Clayman, S. Lippman, H. Goepfert, Genetic heterogeneity in saliva from patients with oral squamous carcinomas: implications in molecular diagnosis and screening. J. Mol. Diagn. 3, 164–170 (2001)
K.J. Livak, T.D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2 (−Delta Delta C(T)) Method. Methods 25, 402–408 (2001)
L. Zhu, H. Chen, D. Zhou, D. Li, R. Bai, S. Zheng, W. Ge, MicroRNA-9 up-regulation is involved in colorectal cancer metastasis via promoting cell motility. Med. Oncol. 29, 1037–1043 (2012)
L. Ma, F. Reinhardt, E. Pan, J. Soutschek, B. Bhat, E.G. Marcusson, J. Teruya-Feldstein, G.W. Bell, R.A. Weinberg, Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 28, 341–347 (2010)
A.B. Hui, A. Lin, W. Xu, L. Waldron, B. Perez-Ordonez, I. Weinreb, W. Shi, J. Bruce, S.H. Huang, B. O’Sullivan, J. Waldron, P. Gullane, J.C. Irish, K. Chan, F.F. Liu, Potentially prognostic miRNAs in HPV-associated oropharyngeal carcinoma. Clin. Cancer Res. 19, 2154–2162 (2013)
K.-i. Kozaki, I. Imoto, S. Mogi, K. Omura, J. Inazawa, Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 68, 2094–2105 (2008)
Y. Khew-Goodall, G.J. Goodall, Myc-modulated miR-9 makes more metastases. Nat. Cell Biol. 12, 209–211 (2010)
S.C. Lin, W.G. Shen, C.J. Liu, MiR-134 expression is oncogenic for oral carcinoma. Oral Oncol. 47, S121–S121 (2011)
C.-J. Liu, W.G. Shen, S.-Y. Peng, H.-W. Cheng, S.-Y. Kao, S.-C. Lin, K.-W. Chang, miR-134 induces oncogenicity and metastasis in head and neck carcinoma through targeting WWOX gene. Int. J. Cancer 134, 811–821 (2014)
E. Elyakim, E. Sitbon, A. Faerman, S. Tabak, E. Montia, L. Belanis, A. Dov, E.G. Marcusson, C.F. Bennett, A. Chajut, D. Cohen, N. Yerushalmi, hsa-miR-191 is a candidate oncogene target for hepatocellular carcinoma therapy. Cancer Res. 70, 8077–8087 (2010)
S. Volinia, G.A. Calin, C.G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R.L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C.C. Harris, C.M. Croce, A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U. S. A. 103, 2257–2261 (2006)
C. Hebert, K. Norris, M.A. Scheper, N. Nikitakis, J.J. Sauk, High moility group A2 is a target for miRNA-98 in head and neck squamous cell carcinoma. Mol. Cancer 6, 5 (2007)
E.V. Barker, N.K. Cervigne, P.P. Reis, R.S. Goswami, W. Xu, I. Weinreb, J.C. Irish, S. Kamel-Reid, microRNA evaluation of unknown primary lesions in the head and neck. Mol. Cancer 8, 127 (2009)
Acknowledgments
The authors would like to acknowledge the financial support of the Queensland Government Smart Futures Fellowship Programme (QGSFF), Queensland Centre for Head and Neck cancer and University of Queensland Diamantina Institute Central Funds. In addition, the authors wish to acknowledge the on-going clinical support from the ENT Department at the Princess Alexandra Hospital in Woolloongabba, Australia. Special thanks goes to Ms Dana Middleton, A/Prof Chris Perry and A/Prof Ben Panizza.
Financial disclosure
None.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
(DOCX 21 kb)
Supplementary Table 2
(DOCX 19 kb)
Supplementary Table 3
(DOCX 29 kb)
Supplementary Table 4
(DOCX 12 kb)
Rights and permissions
About this article

Cite this article
Salazar, C., Nagadia, R., Pandit, P. et al. A novel saliva-based microRNA biomarker panel to detect head and neck cancers. Cell Oncol. 37, 331–338 (2014). https://doi.org/10.1007/s13402-014-0188-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-014-0188-2