
Abstract
Olive pomace is a phytotoxic by-product in the olive oil production. Lignin is a biopolymer present in the olive pomace in remarkable percentages, which has a great variety of potential industrial uses. The extraction of lignin using the ionic liquid triethylammonium hydrogen sulfate resulted in recovery yields as high as 40% of the available lignin in the dry olive pomace. This percentage was obtained after optimizing conditions such as temperature, extraction time, and water content in the ionic liquid. This is the first time such a high percentage of extraction has been achieved when evaluating this type of feedstock. For comparison, two other extraction methods (sulfuric acid and alkaline treatments) were used to assess their extracting performances. Lignin was quantified after developing a rapid, robust, and reliable method by Fourier transform infrared spectroscopy (purity of 101 ± 16%) and characterized by proton nuclear magnetic resonance and gel permeation chromatography. The assay total phenolic content (TPC) revealed high content of phenolic groups (212 ± 26.9 mg of gallic acid equivalents per g of lignin). The high purity and TPC conferred on the extracted lignin a potentially high antioxidant activity. In addition, a 67-fold scale-up extraction of initial mass loading was performed obtaining same results as in the lower scale. Thus, the extraction of lignin using this methodology is expected to mitigate the disposal of the olive pomace and provide certain revenue to the oil mill.

Graphical abstract
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Spain is the largest producer of olive oil in the world (> 1 million tons/year), followed by Italy and Greece [1, 2]. Olive oil is one of the pillars of the Mediterranean diet, which is considered a key factor for a healthy life style. However, the production of olive oil generates large quantities of residues, namely olive pomace. The olive pomace is a slurry composed mainly of water and olive stone and flesh. This concentrated mixture of components is a phytotoxic by-product for the environment due to the variations in the pH, high salinity, and high percentage of organic components such as phenolic and lipidic fractions [3], which confer potential antioxidant and antimicrobial properties to the olive pomace [4, 5]. For all this, our current linear economy based on “make, take, and dispose” does not favor the reuse of such waste and its disposal becomes an emerging environmental problem. Some of the actions undertaken to diminish the environmental impact are the extraction of the oil left in the olive pomace (2–9%) by chemical processes [6], and the use of the remaining olive pomace as a combustion agent in the renewable energy market [7], animal feeding [2], or for composting to produce fertilizers [8]. Nowadays, the disposal of the olive pomace is still a matter of concern.
Olive pomace is a heterogeneous by-product rich in lignin. Lignin is a biopolymer present in the olive flesh and stone, although the latter contains higher levels. Lignin is one of the major constituents in the olive pomace accounting for ca 30% of the dry weight (dw) [2] and is the only renewable source of aromatic biopolymers on earth. Lignin from the olive pomace can be of industrial and/or commercial interest [9, 10]. Nowadays, lignin is used for applications related to agriculture, food industry, bioplastics, biofuel, lubricants and in the field of particles (nanocomposites and nanoparticles) [11].
Recently, triethylammonium hydrogen sulfate ([Et3NH][HSO4]) has been used to process biomass (i.e., deconstruction of lignocellulosic feedstock). This inexpensive ionic liquid exhibits low vapor pressure, high thermal stability and recycling capacity [12]. The effectiveness of [Et3NH][HSO4] has already been proven in other biomass than olive pomace. The yields for the lignin recovery spanned between 30 and 100% depending on conditions of the treatment and the type of substrate [13,14,15].
The determination of these yields is mostly performed by chemical and/or gravimetric methods, which are tedious and highly time-consuming [16]. Hence reliable, robust and rapid methods for the determination of lignin are in great demand. In this respect, spectroscopic techniques are well positioned to analyze lignocellulosic biomass. In particular, Fourier transform infrared spectroscopy (FT-IR) covers the demands for robustness, cost-effectiveness, and high throughput by using the sampling technique attenuated total reflectance (ATR). Nevertheless, the reliable quantification by FT-IR is challenging because of bands often overlap and the direct assignment of band areas to concentration of standards is difficult. To overcome this particular problem statistical models (e.g., principal component and partial least squares regressions) have been used for the quantification of lignin [17,18,19], but simpler studies using single wavenumbers corresponding to a characteristic band of lignin have proven that a reliable lignin determination is possible [20, 21].
In this work, we aimed to maximize for the first time the extraction of lignin from the olive pomace using the ionic liquid [Et3NH][HSO4]. We validated a FT-IR method based on the determination of the content of lignin (%) in different types of biomass. The performance of the optimized extraction was compared with two classical methods for the extraction of lignin: sulfuric acid treatment, to obtain “Klason” lignin, and alkaline treatment with sodium hydroxide (NaOH). The three different lignins were characterized by spectroscopic and chromatographic techniques (FT-IR, nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC)). One step further in this work was the scale up of the lignin extraction (67-fold) and the recycling capacity study of the ionic liquid to verify the applicability of the method in a larger scale.
2 Materials and methods
Olive pomace from three different types of crops: integral (INT), conventional (CONV), and organic (ORG) were supplied in the form of a slurry containing water, olive flesh, and stone by an olive oil producer from “Les Garrigues” (Lleida, Spain). The crops were located in different fields, treatments for olive oil production were not exactly the same, and there could also be differences in the type of olives depending on the crop origin. The production of olive oil from INT and CONV crops was undertaken by using two-phase mills, while for ORG was carried out using a three-phase system. The major difference between the two systems is that the two-phase processing does not add water after pressing the olives and centrifuging [2].
The olive pomace was aliquoted and dried in an oven at 60 °C until constant weight was achieved. The dry olive pomace was ground and sieved through 1 mm pore. Further information about samples and their flesh/stone ratio (Table S1 and Fig. S1), solvents, reagents, instrumentation, synthesis of the ionic liquid (Figs. S2–4), and compositional characterization of the olive pomace can be found in the supporting information.
2.1 Quantification of lignin using FT-IR
The quantification of lignin in the samples (e.g., olive pomace, residues from treatments and other substrates) was done by interpolating the area of the characteristic IR vibration of the aromatic skeleton at 1510 cm−1 [20, 22] against the area of seven standards (10%, 17%, 22%, 37%, 50%, 61%, and 100% of lignin) (Fig. S5). The standards were prepared by mixing in a mortar amounts of commercial kraft lignin and cellulose. The accuracy of the FT-IR method was checked by comparing the results of the percentage of lignin from different samples against the gravimetric method based on the fiber determination [23, 24] (Table S2). The gravimetric method was established as the reference method, and the differences from the FT-IR values were evaluated according to the Bland–Altman analysis [25] and the paired samples t test (Fig. S6 and Table S3, respectively). The precision of the method was evaluated as the coefficient of variation of three measures taken at different time points. The robustness was tested by acquiring the same calibration curve after 3 months and quantifying again the previous samples to assess the deviations of the new percentages. For further details about the method validation, see section 2.1 in the supporting information.
2.2 Ionic liquid, sulfuric acid, and alkaline treatment
Dry olive pomace (300 mg) was mixed with [Et3NH][HSO4]. The ratio of sample/ionic liquid was established as ratio 1:10 by weight [13, 15, 26, 27]. The mixtures were subjected to different extraction conditions combining time (2 h, 4 h, 8 h, 24 h), temperatures (50 °C, 90 °C, 120 °C, and 150 °C), and water content in the ionic liquid (0%, 5%, 20%, 40%, and 60%). Once the extraction time was completed, 8 mL of ethanol was added to reduce the viscosity of the mixture and to facilitate the separation of the sediment and the supernatant (centrifugation for 10 min with a g-force of 1400). The sediment was washed with 4 mL of ethanol four times and kept in the oven at 60 °C overnight. The supernatant was evaporated under vacuum to remove and reuse the ethanol in subsequent extractions. To the supernatant, 25 mL of water was added to precipitate the lignin. The lignin was recovered by centrifugation (same conditions as mentioned above), washed 3 times with 1% formic acid in water and kept in the oven at 60 °C until constant weight. The water from the supernatant was rotavaporated, and the ionic liquid was kept for further extractions in order to investigate its re-extracting effectiveness in future experiments.
The alkaline treatment was carried out according to the published procedure from Sun et al. [28], but without the addition of hydrogen peroxide and increasing temperature to reduce time. The sample was mixed with an aqueous media (pH~11.5 using a solution of 7.5% NaOH (w/volume (v)). The extraction process was performed by magnetic stirring at 120 °C for 2 h. The resulting black liquor was diluted with 4 mL of water and centrifuged as mentioned above. The sediment was washed until neutral pH, and the highly basic supernatant was lowered to pH 6, to precipitate unwanted substances, and finally to pH 2 for lignin precipitation. The lignin was isolated after centrifuging, washed with water 1% formic acid, and dried in the oven until constant weight. The sulfuric acid treatment consisted of mixing olive pomace (or pulp residue) and 72% sulfuric acid (v/v) at a ratio 1:10 (w/v) for 2 h at 40 °C. The solution was adjusted to pH 2 with sodium hydroxide, and the residue was filtered through a glass microfiber filter, thoroughly washed with water 1% formic acid and dried until constant weight.
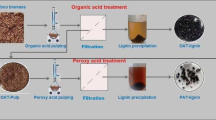
Figure 1 summarizes the most relevant steps of each treatment. The treatment using the ionic liquid did not generate significant waste other than the washing volumes for the lignin, and the ethanol washes from the pulp were recovered by a simple distillation. The sulfuric acid treatment required one filtration less than the other two treatments, and it did not generate pulp, which means that all solid matter, except lignin and coarse particles, was hydrolyzed and diluted in the aqueous medium (filtrate). The alkaline treatment produced up to three solid fractions: water-insoluble substances at pH 6 (sediment 2) and pH 2 (sediment 3 or lignin) and the undissolved pulp (sediment 1).

Schematic flowchart of the sulfuric acid, ionic liquid, and alkaline treatments of the olive pomace
2.3 Determination of the total phenolic content
The total phenolic content (TPC) in lignin was determined by the Folin–Ciocalteau (F-C) assay [29]. Five standards of gallic acid were prepared in dimethylsulfoxide (DMSO) with concentrations ranging from 0.159 to 2.55 mM. Lignin samples and kraft lignin were dissolved in DMSO (0.5 mg/mL and heat when needed). Then, 100 μL of these lignin solutions (also standards and blanks (100 μL of DMSO)) were mixed with 200 μL of a 10% F-C solution in DMSO and 800 μL of a 700 mM sodium carbonate aqueous solution. The samples were kept at room temperature for > 2 h, and the absorbance was measured at 765 nm. The TPC was expressed as mg of gallic acid equivalents (GAE) per gram of lignin.
2.4 Determination of weight average molecular weights
The lignin extracted from the olive pomace was subjected to GPC. A Varian ProStar instrument equipped with a UV-vis detector and two PolarGel-L columns (300 × 7.5 mm) were employed. DMSO with 0.1% lithium bromide (LiBr) was used as the eluent. Samples (~ 4 mg) were dissolved in 5 mL of DMSO (0.1% LiBr), the flow rate was 0.8 mL/min, and the measurements were performed at 50 °C with a detection wavelength at 260 nm. Polystyrene standards (Sigma-Aldrich) ranging from 162 to 19500 g/mol were used for calibrating the system.
3 Results and discussion
3.1 Compositional characterization of the olive pomaces
The three different olive pomaces (i.e., INT, ORG, and CONV) were supplied with the aim to evaluate whether the type of crop could lead to different olive pomaces or not.
The olive pomaces from the oil mill averaged 65 ± 4.0% of water and the flesh/stone ratios were around 1.4 (Table S1). After drying the samples, the most prominent group of constituents was the extractables (41 ± 1.5% dw or 14 ± 0.5% of the wet weight (ww)). From this 41%, oils and waxes accounted for 20 ± 1.1% dw, being the remaining 21 ± 0.7% dw assigned to sugars, pigments, proteins, residual moisture, etc. The extraction of the oils and waxes using organic solvents was regarded as quantitative after two cycles. The presence of oils and waxes in the sample was monitored by FT-IR and NMR. The spectra from both techniques revealed that the majority of the dissolved matter corresponded to triglycerides (Figs. S7–S8). The huge reduction of the characteristic functional groups of the triglycerides in the spectra from the olive pomaces, confirmed the assumed quantitative removal of the triglycerides (Fig. S9).
Lignin was the second most abundant constituent in the olive pomace averaging 37 ± 3.9% dw or 13 ± 1.3% ww. The following components in percentage were cellulose (17 ± 2.0% dw or 6.0 ± 0.7% ww) and hemicellulose (4.4 ± 2.0% dw or 1.5 ± 0.7% ww) (Table 1).
As seen in Table 1, this method gave huge variations in the determination of hemicellulose for INT and ORG samples (coefficients of variation (CV) > 80%; n = 3). Consequently, when using sample weights of 1 g, the results in the determination of relatively low percentages of hemicellulose (e.g., < 5%) must be taken with caution. The rest of the components provided satisfactory CV (< 20%; n = 2–3).
According to the results from Table 1, the compositional determination of the three different types of crops do not actually differ from each other, which means that all olive pomaces can be used as a single sample for their subsequent valorization. Furthermore, the percentages of the compositional analysis are in good agreement with other characterizations of the olive pomace [30].
3.2 Deconstruction of the olive pomace
3.2.1 Ionic liquid treatment
The synthetized ionic liquid [Et3NH][HSO4] contained 20 ± 0.99% (n = 3) of water. Its density was 1.1862 ± 0.0105 g/mL (n = 4; 27 °C) and the pH 0.49 ± 0.09 (n = 2). The use of [Et3NH][HSO4] for the deconstruction of other substrates than olive pomace has recently been reported elsewhere [13,14,15]. The optimized extraction conditions were commonly reported as heating temperatures about 120 °C, extraction time around 4 h, and water content of 20%. We maintained these conditions for the design of our experiments, and we also explored upper and lower ranges to find out the optimal conditions for the lignin extraction from the olive pomace (Fig. 2). The lignin percentages reported in this study corresponded to the lignin recovered with respect to the available lignin in the olive pomace (mean of 37 ± 3.9%; n = 3), unless otherwise specified.

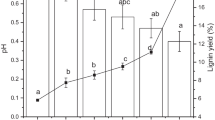
Optimization of the different parameters for the extraction method using [Et3NH][HSO4]. a Lignin recovery after applying different combinations of temperature/time, while keeping the ionic liquid with 20% water. b Lignin recovery after applying different percentages of water in the ionic liquid, while keeping the temperature and time at 120 °C and 4 h, respectively. The black line shows the ratio “solid residue (pulp+lignin)/dissolved matter.” c Percent recovery of the pulp (orange) and lignin (blue) over time, while keeping the temperature and water content at 120 °C and 5%, respectively
The temperature of extraction was a key factor for the good delignification using [Et3NH][HSO4]80%. Extractions at 50 °C for 66 h, and 90 °C for 8 h yielded lignin recoveries lower than 5%. Temperature at 120 °C and 4 h extraction gave a lignin recovery of 31 ± 9.6% (n = 6). Strikingly, a higher temperature (150 °C) yielded only slightly higher recoveries (40 ± 1.1%; n = 2) after extracting for 2 h (Fig. 2a)). Wigand et al. [14] achieved an increment of 100% lignin recovery using the same conditions (150 °C/2 h) in willow (Salix) with particle sizes in the range of 180–850 μm. This huge difference in performance of the same ionic liquid can be attributed mostly to structural composition of the lignin according to the ratio between syringyl and guaiacyl units (hardwood vs softwood). Furthermore, lignin is a cross-linked three-dimensional polymer with variable number of linkages to the structural carbohydrates in the lignocellulosic materials [31]. Therefore, lignocellulosic substrates presenting an elevated number of these linkages will presumably show higher recalcitrance towards the extraction, even after applying harsher conditions [32]. Finally, different physical properties also may influence in the performance. The low capacity of olive stones for swelling in liquid media make them much more recalcitrance than other substrates like wood or straw using the same conditions. One solution to overcome this situation would be to reduce the particle size as much as possible [31]. We tested this hypothesis for one sample from a new batch of olive pomace. The sample was sieved through 1 mm, 0 .5mm, and 0.25 mm pores and extracted according to the optimized method (120 °C/4 h). The lignin recoveries obtained for the different particle sizes were 21.1 ± 1.77% (n = 2), 32.3 ± 1.06% (n = 2), and 39 ± 2.76% (n = 2), respectively. The results showed that reducing the particle size is translated into higher lignin recoveries [33].
Regardless of the lignin recovery, working at elevated temperatures (> 120 °C) may not be advisable because thermal stability issues may arise for the ionic liquid and cellulose, and at the same time undesired side-reactions from the biomass may occur. For example, the generation of pseudo-lignin, which precipitates and reduces the lignin purity, has previously been observed [14, 34].
Extractions using different water content in the ionic liquid were also investigated. Based on the experiments mentioned above and previous findings, temperature of extraction was set at 120 °C for 4 h [27, 33, 35] and the ionic liquid solutions were prepared containing 0%, 5%, 20%, 40%, and 60% of water. At the end of the extractions, three fractions were obtained: a residue (pulp rich in cellulose), a precipitate (mostly lignin), and the used ionic liquid with dissolved matter (Fig. 2b)).
During the extraction, the low content of the amorphous hemicellulose in the olive pomace (< 7% dw) is assumed to be dissolved in the ionic liquid, while dissolution of the cellulose is considered negligible [13]. In fact, we tested the solubility of commercial cellulose in [Et3NH][HSO4]95% 1:10 (w/w) and the recovered amount after applying the extraction conditions mentioned above was 97 ± 11% (n = 2). Triglycerides present in the olive pomace may appear with the lignin after addition of water. Finally, lignin has a clear partitioning as seen in the FT-IR spectra from pulp and precipitate (Fig. S10). The characteristic bands of insoluble triglycerides (stretching vibration (ν) C=C-H 3003 cm−1; νC-H 2923 and 2854 cm−1; and νC=O 1740 cm−1) [20] can be seen in the precipitate (green), and the characteristic band of lignin (νC=C 1510 cm−1) [20, 36] is visible in both, pulp (blue) and precipitate. A more thorough discussion about the FT-IR characterization of lignins obtained from the three different treatments is described in section 3.3.
At this point, the extracted lignin was purified to remove the water insoluble triglycerides. Hand-shaking (twice) of the sample with 1.5–2 mL of acetone (or hexane) was sufficient to substantially remove the triglycerides (Fig. S11). The percentage of triglycerides extracted from two different lignins was 16 ± 2.0% (n = 2). This simple and fast clean-up was evaluated as more efficient than performing the solid–liquid extraction over the olive pomace. The latter extraction required two cycles of 8 mL toluene/ethanol (3:1; v/v) and sonication 50 min each (same procedure using acetone) (Fig. S8) resulting in a pure lignin (104% according to the quantification by FT-IR).
Lignin and pulp recoveries were plotted against varying percentage of water in the ionic liquid (Fig. 2b)). Water contents in the ionic liquid of 0%, 40%, and 60% gave recovery yields lower than 24% (< 9% dw from the total biomass), while the ionic liquid with water content of 20% and 5% gave lignin recoveries of 31 ± 9.6% and 37 ± 4.9%, respectively (11% and 14% dw from the total biomass). Water content below 5% hindered the recovery of lignin and disturbed the increasing tendency seen in Fig. 2b). This is actually in agreement with previous studies were this phenomenon was also observed in ionic liquids based on butylimidazolium hydrogen sulfate [35, 37]. Although the ionic liquids are not exactly the same, the comparison can be regarded as valid because the role of the cation in the solubility of the lignin is much less important than the anion [12].
This is the first time that the ionic liquid [Et3NH][HSO4] is used to extract lignin from olive pomace. After optimization of the extraction methodology, the highest lignin recovery was 44%. Nevertheless, the mean was 30.9% (n = 6; CV = 31%) when considering three experiments (n = 2 each; CV ≤ 23%) performed at different time points. Previous studies have tested the extracting efficiency of [Et3NH][HSO4]80% in substrates like grasses (miscanthus) [13, 27, 38] and hardwood (willow) [14, 15]. The reported lignin recoveries for miscanthus were close to 100% and for willow lower than 40% (with respect to the available lignin content). The content of lignin in miscanthus (gigantheus) and willow was reported to be the same (~ 25% dw from the total biomass), but the degree of delignification in grasses was much more effective [31]. Due to the recoveries obtained for lignin in our samples, one should regard the olive pomace more as hardwood/softwood substrate rather than grass based on lignin recoveries from Fig. 2. As a result of the high lignin recoveries obtained in grasses by other researchers, we tested the efficiency of our [Et3NH][HSO4]80% extracting lignin from alfalfa (8.1% dw lignin from the total biomass). The recovered lignin was 96 ± 0.88% (n = 2) after applying the optimal conditions obtained for the olive pomace.
The pulp remained constant around 54% except when extracting with the ionic liquid containing a water content ≥ 40%, and then this percentage dropped to around 40%. The ratio “total solid residue (lignin+pulp)/dissolved matter” for [Et3NH][HSO4]40% was 0.7, indicating that an important fraction of biomass was more soluble than in the previous conditions (Fig. 2b)). Most of the dissolved matter seemed to be only water soluble since the ratio “total solid residue/dissolved matter” increased up to 1.9 for [Et3NH][HSO4]95% and [Et3NH][HSO4]100%. A likely explanation could be that when having low water contents in the ionic liquid, the water-soluble substances present in the olive pomace saturate the aqueous medium, and therefore, part of them contributes to increase the percentage of pulp. When the system contains more water, those substances are dissolved more easily because the viscosity of the solution is highly reduced and the water content is higher. As a result, there is a reduction of the ratio “total solid residue/dissolved matter.” This is actually in line with the results obtained from the compositional analysis where the determination of extractables (dissolved matter) yielded more than 40% in warm aqueous solution. Extractables from olive pomace have been reported as high as nearly 50% [30]. Furthermore, the acid hydrolysis can also be enhanced with a higher water content. Yet, the conditions using high percentages of water in the ionic liquid are detrimental for the lignin valorization, and thus, the optimal water content in [Et3NH][HSO4] to extract lignin was evaluated between 5 and 20%.
The behavior of the extraction efficiency of the ionic liquid over time was also examined using [Et3NH][HSO4]95% (Fig. 2c)). In general, the longer the extraction time, the more lignin was recovered and more biomass was dissolved. The percentage of pulp after 4 h was 52%, and after 8 h of extraction, it remained steady at 35%. Likewise, lignin experienced a plateau after approximately 4 h of extraction. The increment of lignin recovery from 4 to 24 h was only 15% (of the available lignin in the olive pomace). This increment may not be sufficient to justify the energetic expense and time consuming extraction when thinking to move from lab-scale to pilot- or industrial-scale (for further details see section 3.2.1 in the supporting information). The scale-up of the extraction methodology using 20 g of olive pomace (67-fold higher than the initial mass) was carried out in a 500-mL two-necked round-bottom flask with a condenser. The system was heated in an oil bath at 120 °C. The lignin extraction was highly satisfactory obtaining a recovery of 41.5% from the available lignin in the olive pomace. The lignin was characterized by FT-IR, and all characteristic bands matched with the spectrum from lignin obtained at much lower scale (Fig. S12). In addition, the recovery of the ionic liquid after water removal was almost quantitative (94%; n = 4; CV = 0.6%) and its performance in three subsequent re-extractions of a new batch of olive pomace was highly satisfactory since the recycled ionic liquid did not lose capacity of extraction at all (Table S4). In fact, the re-extractions yielded higher lignin recovery than the initial one with fresh ionic liquid. This phenomenon was also observed in a previous study [13].
The high recovery of the ionic liquid, the good recovery of lignin in our scale-up extraction, as well as, in subsequent uses of the ionic liquid, showed the great potential to incorporate this methodology in a pilot plant.
3.2.2 Alkaline treatment
The alkaline treatment (pH 11.5; 2 h; 120 °C) [28] resulted in apparent lignin recoveries of 24 ± 4.8% (n = 4) (9.1% dw from the total biomass). These values are very similar to those obtained using [Et3NH][HSO4]95% (2 h; 120 °C) (Fig. 2c)). The percentages obtained using this treatment in comparison with the yields reported in the literature were not satisfactory. Although it is difficult to establish a fair comparison due to differences in the treatment conditions, lignocellulosic substrate, content and structure of lignin, our recoveries were much lower than the lignin recovered from sugarcane bagasse (40–72% dw from the available lignin) [39].
Moreover, the FT-IR spectrum of lignin from the alkaline treatment revealed more bands belonging to unwanted substances such as polysaccharides (nonsymmetric bridge C–O–C 1157 cm−1 and nonsymmetric out-phase ring 895 cm−1) [40] (Fig. S13). Consequently, the effect obtained using the alkaline treatment was more an enrichment of lignin rather than an extraction of purer lignin like in the ionic liquid treatment.
On the other hand, the alkaline treatment left much less pulp than the treatment using [Et3NH][HSO4]95% (i.e., 25% vs 52%, respectively). Therefore, the apparent delignification capacity of both treatments was basically the same, but the alkaline treatment dissolved the biomass to a much higher extent than the ionic liquid treatment did. In light of these results, we decided to apply the alkaline treatment to the pulp from the ionic liquid treatment (approximately 0.18 g) in order to dissolve undesired components and increase the enrichment of cellulose in the pulp. Prior to combine the treatments, we performed re-extractions of lignin from the pulp left after the ionic liquid treatment using fresh ionic liquid. Likewise, we undertook another experiment doing the same with a solution of NaOH (7.5% w/v). The second and consecutive ionic liquid treatments resulted in lignin recoveries lower than 8%, and for the alkaline treatment, the lignin precipitation at pH 2 simply did not happen. This indicated the recalcitrant nature of the remaining lignin towards these treatments [13].
After applying the ionic liquid and alkaline treatment sequentially to the olive pomace, no lignin was recovered from the black liquor in the second treatment, but the pulp accounted for 18 ± 1.8% (n = 2) of the total biomass used for the experiment. This percentage basically corresponded to the theoretical amount of cellulose in the olive pomace (see Table 1). However, the spectrum of the pulp showed a solely mixture of cellulose and lignin (Fig. S14). The standard containing 63% cellulose and 37% kraft lignin was compared with the pulp. All bands matched between both spectra and the lignin content by FT-IR indicated 40% by weight; thus, the percentage of cellulose was presumably 60%. Nevertheless, the content of cellulose in the pulp was determined indirectly by acid hydrolysis (72% sulfuric acid; 40 °C; 2 h). This is an effective method for the total solubilization of the cellulose (see below). The percentage of lignin determined gravimetrically was 34 ± 1.9% (n = 2). Again, the persistent occurrence of lignin after applying two treatments may be attributed to the steric hindrance of the reagents to break the lignin carbohydrate linkage [41]. Therefore, the cellulose content in the pulp was assumed to be 66 ± 1.9% (n = 2). This means that the cellulose enrichment of the initial dry olive pomace (17% dw) was increased by a factor of nearly 4. Therefore, the pulp obtained after the first treatments was not used for further lignin extraction, but with a subsequent alkaline treatment, the pulp could be further exploited for a saccharification process.
3.2.3 Sulfuric acid treatment
The sulfuric acid treatment (72% v/v) was used to recover lignin since most of the biomass and in particular any polysaccharide will be hydrolyzed at such acidic conditions [42]. Therefore, after the treatment with sulfuric acid only lignin, char and minerals were expected to remain as solids [43]. The lignin recovery in the olive pomace was 38 ± 3.2 (n = 4), which represents 103% of the total lignin in the olive pomace (the purity of lignin after removing the triglycerides was 101 ± 16% by FT-IR). Recovery percentages higher than 100% using H2SO4 have been previously reported [44]. The presence of minerals and or other thermally resistant substances in a substantial percentage was disregarded due to the very low ash content of the extracted lignin (0.34 ± 0.09%; n = 3).
3.3 FT-IR and NMR spectroscopic characterization of lignin
Table 2 shows the wavenumbers of the most relevant vibrational IR bands, and Fig. 3 shows the FT-IR spectra of the extracted lignin using the ionic liquid, sulfuric acid, and alkaline treatments.

Spectra of lignin recovered using the alkaline (green), ionic liquid (blue), and sulfuric acid (brown) treatments after applying the clean-up with acetone
The spectrum of the lignin from the alkaline treatment was somewhat different because of the cellulose content. Band 1 (ν O-H) appeared more intense than in the other spectra because of the likely contribution of the hydroxyl groups of the cellulose. Likewise, bands 9 and 10 (C–O and nonsymmetric out-phase cellulose ring, respectively) evidenced the presence of cellulose.
The spectra of “purer” lignins from the sulfuric acid and ionic liquid treatments showed a more similar match among bands. Bands 1 (ν OH), 2 and 3 (ν C-H), 7 (aromatic skeletal vibration), and 8 (asymmetric δ C–H) fell in the same wavenumbers, and only band 4 (nonconjugated carbonyl) in the sulfuric acid treatment was slightly shifted towards higher frequencies (i.e., 1719 cm−1). More specifically, the band at 1719 cm−1 is attributed to the esterification of the phenol and alcohol of the propane chain [50]. This latter band corresponding to C=O stretching was also observed in solvolysis lignin [20]. The FT-IR band close to 1700 cm−1 has also been found in hardwood and softwood (Eucalyptus globulus and Pinus radiata, respectively) [51]. However, certain type of lignins like commercial kraft lignin showed a total absence of bands 4 and 5 (Fig. S15).
A more remarkable difference between these two spectra resided in the carbonyl/aromatic region (i.e., bands 4, 5, and 6), in addition to the finger print region below 1200 cm−1. Band 6 (aromatic skeletal vibration) from lignin extracted using the ionic liquid treatment is absent in the sulfuric acid treatment. During the sulfuric acid treatment, lignin has suffered a structural change or chemical reorganization in the structure, which has resulted in an intense band corresponding to the band 5 (absorbed O–H and/or conjugated C=O), which is absent in the lignin obtained from the ionic liquid treatment. Since the water content in the lignins from the acid treatments is negligible, we ascribed this band only to the conjugated C=O stretching. Therefore, the oxidative process experienced in the sulfuric acid treatment is higher than in the ionic liquid treatment. This can also be seen qualitatively in the relative decrease in intensity of the band at 1511 cm−1 compared to the band at 1719 cm−1, which indicated the decomposition of aromatic structures. Both bands (5 and 6) have been reported together in LignoBoost lignin [36].
Bands 4, 5, and 6 were also detectable in the alkaline treatment. The frequency of bands 4 and 6 has almost a perfect match with the respective wavenumbers belonging to lignin from the ionic liquid treatment (discussed above). Band 5 may correspond not only to the conjugated C=O vibration but also to the absorbed OH [47]. Since the OH stretching signal at 3372 cm−1 is prominent in the alkaline treatment, we assumed that the major contribution should come from the absorbed OH. Both FT-IR signals corresponding to OH vibrations have also been seen in the spectrum of the wet ionic liquid (Fig. S2) and both disappeared in the spectrum of the dry ionic liquid (Fig. S4).
Regarding the NMR experiments, only 1H-NMR spectra gave useful information for the characterization of the extracted lignin (without clean-up). The combination of the available amount of sample, the type of probe, and the sensitivity of the NMR instrument were not sufficient to obtain meaningful results from the 13C-NMR experiments. Table 3 shows the relative abundance of integrated protons from the 1H-NMR spectra (Figs. S16–18). The protons were divided in three groups: carboxylic (13–10 ppm) phenolic (10–7.5 ppm) aromatic units (7.5–5.5 ppm), oxygenated aliphatic (o-aliphatic) side chain (5.5–2.5 ppm), and aliphatic side chain (2.5–0 ppm).
The high percentages of aliphatic side chains in the three treatments are associated with aliphatic protons from the triglycerides or triglycerides transformed into fatty acids. The presence of triglycerides/fatty acids was confirmed by their characteristic signals >CH–O–CO–R at 5.1 ppm and –HC=CH– at 5.3 ppm (Figs. S16–18). The higher phenolic and aromatic proton percentage corresponded to the lignin from the ionic liquid treatment, which is in accordance with the FT-IR evidences and with the highest total phenolic content assay (see below). In addition, the lignin obtained from the scale-up extraction using the ionic liquid treatment only differed in the percentage of the o-aliphatic (34%) and aliphatic protons (56%).
The sulfuric acid treatment presented similar values for the side chain protons, but the phenolic percentage decreased dramatically as well as the aromatic units. This can be attributable most likely to condensation, re-polymerization, char formation processes [13, 14, 35], and/or aromatic proton depletion as evidenced in the FT-IR.
The alkaline treatment presented also lower percentage of aromatic and phenolic protons, although the purity, as mentioned above, was much lower (66% by FT-IR) than the lignin from the ionic liquid treatment. The percentage in the o-aliphatic region was significantly higher due to the proton contribution of the polysaccharides present in the sample (between 5 and 3 ppm) [52]. Moreover, the higher percentage of carboxylic protons than in the two acid treatments could be explained by the faster hydrolysis of the triglycerides into fatty acids under the basic conditions of the alkaline treatment.
3.4 Total phenolic content and weigh average molecular weights
The measurement of the TPC in the three different type of lignins revealed that the highest % GAE was obtained for the lignin extracted using the ionic liquid (212 ± 26.9 mg GAE/g lignin) followed by the alkaline treatment (97.1 ± 26.8 mg GAE/g lignin) and the sulfuric acid treatment (41.1 ± 18.9 mg GAE/g lignin). For comparison, TPC was measured to commercial kraft lignin (204 ± 77.6 mg GAE/g lignin).
The TPC results suggested that the ionic liquid treatment preserved the phenolic units of the polymer providing more likely better antioxidant capacity than the other two types of lignin. This is in line with the higher percentage of phenolic protons determined by NMR (Table 3). For the sulfuric acid treatment, the solubilization of the 2–4 mg was incomplete due to most probably cross-linking of the lignin caused by the harsh acidic conditions of the treatment [14], and thus, the TPC value was the lowest. In addition, the TPC could be expected lower because of the probable oxidation processes seen in the FT-IR (Fig. 3). For the alkaline treatment, all the samples were dissolved but the reduced TPC could be ascribed to the lower purity of lignin observed in the FT-IR (66% pure lignin by FT-IR). If the purity correction is performed, the TPC increases to 147 mg GAE/g lignin, a value still lower than the TPC obtained with the ionic liquid, but also in agreement with the lower phenolic protons seen in the NMR experiment.
The only treatment giving TPC in the range of the Kraft lignin was the ionic liquid treatment (both around 200 mg GAE/g lignin) [53]. Therefore, the lignin recovered using the ionic liquid treatment can be considered of higher quality in terms of purity (FT-IR and NMR) and TPC with respect to the other two treatments.
The weigh average molecular weight (Mw) determination was regarded as qualitative because of two main reasons: (i) surprisingly, the lignin polymer spanned beyond the upper limit of our GPC system (60,000 Da), and (ii) the solubility of our highest molecular weight polystyrene standard (50,000 Da) in DMSO was too poor and our final calibration range spanned from 162 to 19,050 Da. In any case, the main scope of the GPC measurement was to compare differences among the three treatments and this assessment can be conducted with the data acquired. Both lignins from the ionic liquid treatment (multi and milligram scale) presented approximately 40% lower Mw than the other two treatments (Fig. S19) and the polydispersity (> 3) indicated a broad range of different polymers in all samples. This indicated again that the extraction of lignin using the ionic liquid treatment gave better results in comparison to the other two treatments.
4 Conclusions
The simple, fast, and robust determination of lignin was achieved by using a FT-IR spectrometer equipped with the ATR sampling technique. The correlation between the reference method (gravimetric determination of “Klason” lignin) and the percentages obtained from the areas of the band 1510 cm−1 was high (R = 0.95), and the statistical analyses concluded that there were no significant differences between the percentages reported using either methods.
The ionic liquid [Et3NH][HSO4]80–95% showed good performance extracting lignin from olive pomace and for the first time percentages of extraction near 40% have been reported for this substrate. Lignin recovery increases when particle size of the olive pomace and the flesh/stone ratio decreases.
The satisfactory results of the scale-up extraction pave the way to incorporate this methodology at a pilot scale. In addition, the ionic liquid treatment offered lower Mw polymer, higher TPC, and more relative percentage contribution of the phenolic and aromatic protons by 1H-NMR than the sulfuric acid and alkaline treatments.
References
International Olive Council. World olive oil figures. http://www.internationaloliveoil.org/estaticos/view/131-world-olive-oil-figures. Accessed 18 Dec 2018
Molina-Alcaide E, Yáñez-Ruiz DR (2008) Potential use of olive by-products in ruminant feeding: a review. Anim Feed Sci Technol 147:247–264. https://doi.org/10.1016/j.anifeedsci.2007.09.021
Sierra J, Martí E, Garau MA, Cruañas R (2007) Effects of the agronomic use of olive oil mill wastewater: field experiment. Sci Total Environ 378:90–94. https://doi.org/10.1016/j.scitotenv.2007.01.009
Cicerale S, Lucas L, Keast R (2010) Biological activities of phenolic compounds present in virgin olive oil. Int J Mol Sci 11:458–479. https://doi.org/10.3390/ijms11020458
Cicerale S, Conlan X, Sinclair A, Keast R (2009) Chemistry and health of olive oil phenolics. Crit Rev Food Sci Nutr 49:218–236. https://doi.org/10.1080/10408390701856223
International Olive Council. Quality management guide for the olive-pomace oil extraction industry. http://www.internationaloliveoil.org/estaticos/view/222-standards. Accessed Feb 2019
Missaoui A, Bostyn S, Belandria V, Cagnon B, Sarh B, Gökalp I (2017) Hydrothermal carbonization of dried olive pomace: energy potential and process performances. J Anal Appl Pyrolysis 128:281–290. https://doi.org/10.1016/j.jaap.2017.09.022
Fernández-Hernández A, Roig A, Serramiá N, Civantos CGO, Sánchez-Monedero MA (2014) Application of compost of two-phase olive mill waste on olive grove: effects on soil, olive fruit and olive oil quality. Waste Manag 34:1139–1147. https://doi.org/10.1016/j.wasman.2014.03.027
Ma R, Xu Y, Zhang X (2015) Catalytic oxidation of biorefinery lignin to value-added chemicals to support sustainable biofuel production. ChemSusChem 8:24–51. https://doi.org/10.1002/cssc.201402503
Wang S, Shuai L, Saha B et al (2018) From tree to tape: direct synthesis of pressure sensitive adhesives from depolymerized raw lignocellulosic biomass. ACS Cent Sci 701–708. https://doi.org/10.1021/acscentsci.8b00140
Norgren M, Edlund H (2014) Lignin: recent advances and emerging applications. Curr Opin Colloid Interface Sci 19:409–416. https://doi.org/10.1016/j.cocis.2014.08.004
Hart WES, Harper JB, Aldous L (2015) The effect of changing the components of an ionic liquid upon the solubility of lignin. Green Chem 17:214–218. https://doi.org/10.1039/C4GC01888E
Brandt-Talbot A, Gschwend FJV, Fennell PS, Lammens TM, Tan B, Weale J, Hallett JP (2017) An economically viable ionic liquid for the fractionation of lignocellulosic biomass. Green Chem 19:3078–3102. https://doi.org/10.1039/C7GC00705A
Weigand L, Mostame S, Brandt-Talbot A et al (2017) Effect of pretreatment severity on the cellulose and lignin isolated from Salix using ionoSolv pretreatment. Faraday Discuss 00:1–19. https://doi.org/10.1039/C7FD00059F
Prado R, Erdocia X, De Gregorio GF et al (2016) Willow lignin oxidation and depolymerization under low cost ionic liquid. ACS Sustain Chem Eng 4:5277–5288. https://doi.org/10.1021/acssuschemeng.6b00642
Sluiter A, Hames B, Ruiz R et al (2012) Determination of structural carbohydrates and lignin in biomass. In: Natl Renew Energy Lab. https://www.nrel.gov/docs/gen/fy13/42618.pdf. Accessed 26 Feb 2019
Le DM, Nielsen AD, Sørensen HR, Meyer AS (2017) Characterisation of authentic lignin biorefinery samples by Fourier transform infrared spectroscopy and determination of the chemical formula for lignin. Bioenergy Res 10:1025–1035. https://doi.org/10.1007/s12155-017-9861-4
Zhou G, Taylor G, Polle A (2011) FTIR-ATR-based prediction and modelling of lignin and energy contents reveals independent intra-specific variation of these traits in bioenergy poplars. Plant Methods 7:1–10. https://doi.org/10.1186/1746-4811-7-9
Silva JC e, Nielsen BH, Rodrigues J et al (1999) Rapid determination of the lignin content in Sitka spruce (Picea sitchensis (Bong.) Carr.) Wood by Fourier transform infrared spectrometry. Holzforschung 53:597–602. https://doi.org/10.1515/HF.1999.099
Bui NQ, Fongarland P, Rataboul F, Dartiguelongue C, Charon N, Vallée C, Essayem N (2015) FTIR as a simple tool to quantify unconverted lignin from chars in biomass liquefaction process: application to SC ethanol liquefaction of pine wood. Fuel Process Technol 134:378–386. https://doi.org/10.1016/j.fuproc.2015.02.020
Saad S, Issa R, Fahmy M (1980) Infrared spectroscopic study of bagasse and unbleached high-yield soda bagasse pulps. Holzforschung 34:218–222. https://doi.org/10.1515/hfsg.1980.34.6.218
Rodrigues J, Faix O, Pereira H (1998) Determination of lignin content of Eucalyptus globulus wood using FTIR spectroscopy. Holzforschung 52:46–50. https://doi.org/10.1515/hfsg.1998.52.1.46
Van Soest PJ, Robertson JB (1985) Analysis of Forages and Fibrous Foods, vol 613. Cornell University, Ithaca, pp 202
Van Soest P, Robertson J, Lewis B (1991) Methods for dietary fiber, detergent fiber, and non-starch polysaccharides in relation to animal nutrition. J Dairy Sci 74:3583–3597. https://doi.org/10.3168/jds.S0022-0302(91)78551-2
Giavarina D (2015) Understanding Bland Altman analysis. Biochem Med 25:141–151. https://doi.org/10.11613/BM.2015.015
Weerachanchai P, Lee JM (2017) Recovery of lignin and ionic liquid by using organic solvents. J Ind Eng Chem 49:122–132. https://doi.org/10.1016/j.jiec.2017.01.018
Prado R, Brandt A, Erdocia X, Hallet J, Welton T, Labidi J (2016) Lignin oxidation and depolymerisation in ionic liquids. Green Chem 18:834–841. https://doi.org/10.1039/C5GC01950H
Sun R, Tomkinson J, Wang S, Zhu W (2000) Characterization of lignins from wheat straw by alkaline peroxide treatment. Polym Degrad Stab 67:101–109. https://doi.org/10.1016/S0141-3910(99)00099-3
Ainsworth E, Gillespie K (2007) Estimation of total phenolic content and otheroxidation substrates in plant tissues using Folin-Ciocalteu reagent. NatProtoc 2:875–877. https://doi.org/10.1038/nprot.2007.102
Manzanares P, Ruiz E, Ballesteros M et al (2017) Residual biomass potential in olive tree cultivation and olive oil industry in Spain: valorization proposal in a biorefinery context. Span J Agric Res 15:1–12. https://doi.org/10.5424/sjar/2017153-10868
Brandt A, Gräsvik J, Halletta JP, Welton T (2013) Deconstruction of lignocellulosic biomass with ionic liquids. Green Chem 15:550–583. https://doi.org/10.1039/C2GC36364J
Kumar L, Arantes V, Chandra R, Saddler J (2012) The lignin present in steam pretreated softwood binds enzymes and limits cellulose accessibility. Bioresour Technol 103:201–208. https://doi.org/10.1016/j.biortech.2011.09.091
Pinkert A, Goeke DF, Marsh KN, Pang S (2011) Extracting wood lignin without dissolving or degrading cellulose: investigations on the use of food additive-derived ionic liquids. Green Chem 13:3124. https://doi.org/10.1039/c1gc15671c
Sannigrahi P, Kim DH, Jung S, Ragauskas A (2011) Pseudo-lignin and pretreatment chemistry. Energy Environ Sci 4:1306–1310. https://doi.org/10.1039/C0EE00378F
Verdía P, Brandt A, Hallett JP, Ray MJ, Welton T (2014) Fractionation of lignocellulosic biomass with the ionic liquid 1-butylimidazolium hydrogen sulfate. Green Chem 16:1617. https://doi.org/10.1039/c3gc41742e
Abdelaziz OY, Hulteberg CP (2017) Physicochemical characterisation of technical lignins for their potential valorisation. Waste Biomass Valoriz 8:859–869. https://doi.org/10.1007/s12649-016-9643-9
Brandt A, Ray MJ, To TQ et al (2011) Ionic liquid pretreatment of lignocellulosic biomass with ionic liquid–water mixtures. Green Chem 13:2489. https://doi.org/10.1039/c1gc15374a
Gschwend FJV, Brandt A, Chambon CL et al (2016) Pretreatment of lignocellulosic biomass with low-cost ionic liquids. J Vis Exp 1–18. https://doi.org/10.3791/54246
Arni SA (2018) Extraction and isolation methods for lignin separation from sugarcane bagasse: a review. Ind Crop Prod 115:330–339
Poletto M, Ornaghi Júnior HL, Zattera AJ (2014) Native cellulose: structure, characterization and thermal properties. Materials (Basel) 7:6105–6119. https://doi.org/10.3390/ma7096105
Chen S, Ling Z, Zhang X, Kim YS, Xu F (2018) Towards a multi-scale understanding of dilute hydrochloric acid and mild 1-ethyl-3-methylimidazolium acetate pretreatment for improving enzymatic hydrolysis of poplar wood. Ind Crop Prod 114:123–131. https://doi.org/10.1016/j.indcrop.2018.02.007
Fan L, Gharpuray M, Lee Y (1987) Cellulose hydrolysis. Springer
Patil PT, Armbruster U, Richter M, Martin A (2011) Heterogeneously catalyzed hydroprocessing of organosolv lignin in sub- and supercritical solvents. Energy Fuel 25:4713–4722. https://doi.org/10.1021/ef2009875
Shimizu FL, Monteiro PQ, Ghiraldi PHC, Melati RB, Pagnocca FC, Souza W, Sant’Anna C, Brienzo M (2018) Acid, alkali and peroxide pretreatments increase the cellulose accessibility and glucose yield of banana pseudostem. Ind Crop Prod 115:62–68. https://doi.org/10.1016/j.indcrop.2018.02.024
Kia L, Jawaid M, Ariffin H, Alothman OY (2017) Isolation and characterization of microcrystalline cellulose from roselle fibers. Int J Biol Macromol 103:931–940. https://doi.org/10.1016/j.ijbiomac.2017.05.135
García A, González Alriols M, Spigno G, Labidi J (2012) Lignin as natural radical scavenger. Effect of the obtaining and purification processes on the antioxidant behaviour of lignin. Biochem Eng J 67:173–185. https://doi.org/10.1016/j.bej.2012.06.013
Ayoub A, Venditti RA, Pawlak JJ, Sadeghifar H, Salam A (2013) Development of an acetylation reaction of switchgrass hemicellulose in ionic liquid without catalyst. Ind Crop Prod 44:306–314. https://doi.org/10.1016/j.indcrop.2012.10.036
Faix O, Beinhoff O (1988) FTIR spectra of milled wood lignins and lignin polymer models (DHP’s) with enhanced resolution obtained by deconvolution. J Wood Chem Technol 8:502–522
Naumann A, Peddireddi S, Kües U, Polle A (2007) In: Kües U (ed) Wood production, wood technology, and biotechnological impacts. Universitätsveralg Göttingen, pp 179–196
Watkins D, Nuruddin M, Hosur M, Tcherbi-Narteh A, Jeelani S (2015) Extraction and characterization of lignin from different biomass resources. J Mater Res Technol 4:26–32. https://doi.org/10.1016/j.jmrt.2014.10.009
Casas A, Alonso MV, Oliet M, Rojo E, Rodríguez F (2012) FTIR analysis of lignin regenerated from Pinus radiata and Eucalyptus globulus woods dissolved in imidazolium-based ionic liquids. J Chem Technol Biotechnol 87:472–480. https://doi.org/10.1002/jctb.2724
Dutta T, Papa G, Wang E, Sun J, Isern NG, Cort JR, Simmons BA, Singh S (2018) Characterization of lignin streams during bionic liquid-based pretreatment from grass, hardwood, and softwood. ACS Sustain Chem Eng 6:3079–3090. https://doi.org/10.1021/acssuschemeng.7b02991
do Santos PSB, Erdocia X, Gatto DA, Labidi J (2014) Characterisation of Kraft lignin separated by gradient acid precipitation. Ind Crop Prod 55:149–154. https://doi.org/10.1016/j.indcrop.2014.01.023
Acknowledgements
The authors would like to thank the Catalan Government for the quality accreditation given to their research group 2017 SGR 828. This work has been partially funded by the Spanish government (CTQ2015-70982-C3-1-R, MINECO/FEDER). E.C. would like to thank “Banco Santander” for Grant X15016 in the framework of the “UdL Impuls” project. The “Cooperativa de l’Albi” is greatly acknowledged for fruitful discussions and kindly providing all olive pomaces.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
Enclosed in the supporting information there are complementary discussions, the FT-IR method validation, figures (Fig. S1–19) corresponding to ionic liquid, lignin and triglycerides spectra (FT-IR and NMR) and GPC determinations, and tables (Table S1–4) of the ratios flesh/stone, lignin content of the substrates determined by FT-IR and reference method, the T test: paired two samples for means, and the ionic liquid recovery and the effectiveness of the re-extractions. (DOCX 1730 kb)
Rights and permissions
About this article

Cite this article
Cequier, E., Aguilera, J., Balcells, M. et al. Extraction and characterization of lignin from olive pomace: a comparison study among ionic liquid, sulfuric acid, and alkaline treatments. Biomass Conv. Bioref. 9, 241–252 (2019). https://doi.org/10.1007/s13399-019-00400-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13399-019-00400-w