
Abstract
In this study, we report for the first time application of magnetic ionic liquid (MIL), (Z)-octadec-9-en-1-aminium tetrachloroferrate (III) in the simultaneous preconcentration and quantification of 3 different categories of pharmaceutical products clomipramine, ketoprofen and loperamide from wastewater and human urine samples using dispersive micro-solid phase extraction detected and quantified in high-performance liquid chromatography with a diode-array detector. The MIL was characterized by magnetic property measurement systems, Fourier-transform infrared spectroscopy, zeta potential, nuclear magnetic resonance, Raman scattering spectroscopy, scanning electron microscopy and thermogravimetric analysis. First, a specified amount of the sorbent was successfully added to the sample solution containing the analytes. Afterward, the sample pH is adjusted and stirred at a constant rate of 700 rpm for 45 min. After the adsorption of the analytes, the sample is then sonicated for desorption then centrifuged and decanted for analysis. After optimization, the limit of detection (LOD) was within 2.3–2.9 μg/L and the linearity range was within 1–750 μg/L for all the analytes. The correlation coefficients (R2) obtained were in the range of 0.9967–0.9974, and the relative standard deviation (RSD, %) for intra-day and inter-day was within 2.7–3.3 and 2.6–3.7, respectively. Finally, the applicability of the method on real wastewater and human urine samples was examined and the preconcentration and quantification of the drugs at microgram levels were confirmed with satisfactory results.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Sample preparation is a crucial stage in pharmaceutical product analysis, because it reduces matrix influence, enhances selectivity and sensitivity as well as column protection [1, 2]. Liquid–liquid is one of the classical analytical techniques used for sample preparation. However, it has major drawbacks, such as excessive use of toxic organic solvents, time-consuming, cleaning, and evaporation to preconcentrate the analytes for analytical detection. To overcome these challenges, dispersive liquid–liquid microextraction (DLLME) was developed which is simple, low cost, better enrichment factor, excellent recovery, and also uses micro-amounts of solvent [3,4,5,6]. However, DLLME is not efficient when dealing with complex matrices and not suitable for solvents with higher densities than water [7]. Since then, there has been continuous search for improved methods of sample preconcentration, such as the polypropylene porous membrane protected micro-solid phase extraction [8], solid-phase microextraction [9, 10], dispersive solid-phase microextraction (D-SPME) [11], hollow fiber liquid-phase microextraction (HF-LPME) [12], solid bar microextraction (SBME) [13, 14], effervescent-assisted liquid-phase microextraction (EA-LME) [15], sorbent packed device [16, 17], headspace solid‐phase microextraction [18], molecular imprinting-based solid-phase extraction [19, 20], magnetic molecularly imprinted nanoparticles [21], Dual-template molecularly imprinted polymers [22], etc. All these techniques have been useful for various applications; however, dispersive micro-solid phase extraction (D-µSPE) is preferred due to higher efficiency, simplicity, etc. [23].
Although the D-µSPE is appealing to many scholars, the choice of correct sorbent for preconcentration of targeted analytes remains a big challenge. However, many researchers demonstrated novel sorbents with excellent scientific value in sensitive, selective and detection of various analytes [24,25,26,27,28,29,30,31,32,33]. In recent years, environmentally friendly ionic liquids (ILs) have gained tremendous attention for applications in numerous research areas, including analytes preconcentrations. The ILs are low melting point salts that are characterized with high thermal stability, low vapor pressure, high viscosity, and excellent reusability [34]. Lately, magnetic ionic liquids (MILs) have emerged to be promising sorbent for analytical applications because of the ease of separation from sample matrix by the use of strong external magnet [35]. Our group has recently reported iron-containing ionic liquids for the microextraction of 19 organochlorine pesticides and adsorption of cationic and anionic dyes [36, 37]. Thus, this study uses the advantages of MILs as a sorbent at room temperature for the extraction of three different types of drugs from wastewater and human urine samples.
Emerging contaminants are becoming a major concern in both private and public sectors particularly, in water resources. Among such contaminants are different types of drugs that get into the water body through wide use by humans and animals. One such drug is Clomipramine (CLOM), tricyclic antidepressant, used for the treatment of several disorders. It is one of the most likely pharmaceutical substances that can be found in the environment [38]. It undergoes various transformational stages, but it is mainly metabolized in the liver into desmethylclomipramine via the demethylation process and 66% of it is excreted from the body through the urine. Recently, the drug gained interest in the fields of forensic and clinical studies due to its high risk of suicidal incidents [39]. Loperamide (LOP) which is also administered to patients to reduce diarrhea, short bowel syndrome as well as other inflammatory stomach upset is another concern in the water bodies. The drug and its metabolites are largely released from the body through the feces ending up in the wastewater. It is also reported to be one of the most abused drugs causing a higher concentration of the compounds in the wastewater disposal plants [40]. Ketoprofen (KET) is another pharmaceutical contaminant widely used in nonsteroidal anti-inflammatory drugs (NSAIDs) which has both antipyretic and analgesic effects as commonly utilized for human and veterinary medicine. It is associated with side effects, such as blood coagulation, gastric irritation as well as liver and kidney toxicity. In addition, it is utilized by abattoirs at high doses before slaughtering to deceive buyers as the meat appears pale and healthy, however, risking the life of the consumers [41]. The main problem for these drugs in the environment is that they exist at trace level concentration and creating reliable analytical techniques preconcentrating and detecting them remains paramount [42].
The commonly used technique for the analysis of pharmaceuticals and biomedical compounds is high-performance liquid chromatography (HPLC), because it has attainable simultaneous and excellent precision, specificity as well as accuracy. It is usually equipped with UV and photodiode array (PDA) detectors with a wide range of scanning wavelengths [43].
Recently, our group has demonstrated the superiority of (Z)-octadec-9-en-1-aminium tetrachloroferrate (III) ([OA]FeCl4) over (Z)-octadec-9-en-1-aminium trichlorocobaltate (II) in the extraction of carbamazepine [44]. Therefore, in this work, the [OA]FeCl4 was further employed as sorbent for simultaneous extraction of three pharmaceutical drugs CLOM, KET, and LOP from wastewater and human urine samples. The MIL has the advantage of ease preparation and excellent extraction capacity without any modification or functionalization besides shorter retention time of the method. However, the magnetic property of the MIL is not strong for the phase separation but that can be explored and improved in future studies.
2 Materials and Methods
2.1 Materials
Analytical grade solvents, such as acetonitrile, methanol, ethanol, and acetone, were received from Sigma Aldrich. Other chemicals obtained from the same company are hydrochloric acid (HCl), sodium hydroxide (NaOH), sodium chloride (NaCl), oleylamine, and deuterated chloroform (CHCl3). Ferric chloride (FeCl3) was acquired from Fluka Analytical and the standards of the analytical drugs were received from Merck. Phosphoric acid and potassium phosphate monobasic were also purchased from Sigma Aldrich.
2.2 Sample Preparation Procedure
First, 100 ppm stock solution consisting of the three drug analytes was prepared and stored at 4 °C. The wastewater and human urine were sampled in clean containers. The urine sample was obtained from volunteering healthy person who was not given the drugs under this study and afterward stored at -20 °C. The urine sample is first diluted at a ratio of 1:9 v/v with double distilled water after strong centrifugation for 5 min at 4000 rpm. A similar approach was adopted for the wastewater samples but without dilution but filtered with a filter paper to eliminate the suspended solid particles and stored at 4 °C. All samples were adjusted to the optimized pH 7 before any extraction procedure was conducted.
2.3 Instrumentation and Chromatographic Conditions
The separation and the quantification of the analytes were conducted on HPLC Agilent USA 1260 equipped with an autosampler, detector DAD, and separation column of G1315D, G1315D, and XDB-C18 with length 4.6 × 250 mm of 5 μm, respectively. The system was controlled and operated through Agilent technologies Chemstation software. The mobile phase of the instrument consisted of double-distilled water, acetonitrile, and a phosphate buffer solution at a ratio of 50%, 45%, and 5% respectively. The flow rate was maintained throughout at 1 mL/min in all the runs with isocratic mode, sample injection volume of 20 µL and 230 nm UV wavelength.
2.4 Synthesis of MIL [OA]FeCl4
The synthesis of [OA]FeCl4 was performed based on our previously reported procedure [44]. First, 5 mL of the parent amine compound, oleylamine, was mixed with 5 mL of ethanol and treated with an excess amount of 1 M HCl acid dropwise. The mixture was stirred vigorously with a strong magnetic stirrer for 3.5 h and the solvent evaporated using a rotary evaporator to form the intermediate quaternary (Z)-octadec-9-en-1-aminium chloride ([OA]Cl). Afterward, [OA]Cl and FeCl3 were mixed at an equimolar ratio and stirred for 30 h in ethanol. Subsequently, the solvent was removed by a rotary evaporator. The final dark sticky material was extensively washed with double distilled water to neutral pH and lyophilized and stored for 72 h in an oven at around 70 °C to remove any remains of the solvent and water droplets. The synthesis reactions are illustrated below (Scheme 1).

Synthesis reactions for the formation of MIL [OA]FeCl4
2.5 The D-µSPE Procedure
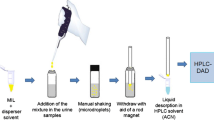
A sample solution of 10 mL containing a known concentration of the three drug analytes is prepared in a 50 mL capped bottle and 40 mg of the synthesized [OA]FeCl4 sorbent added afterward. The pH was adjusted to 3 and the mixture was subjected to constant magnetic stirring for 45 min at 700 rpm for proper dispersion of MIL and diffusion of analytes to its surface for their extraction. After extraction, the analytes-rich sorbents were isolated from the aqueous solution by centrifuging at a speed of 7000 rpm and the aqueous phase was carefully decanted. The analytes were desorbed with 0.4 mL of acetonitrile in an ultrasonic water bath for 45 min and the sorbent was separated by centrifuging at the same speed. The acetonitrile after desorption was loaded in HPLC vials for analysis. A step-by-step D-µSPE procedure is illustrated in (Scheme 2).

Dispersive micro-solid phase extraction procedure
3 Results and Discussion
The MIL [OA]FeCl4 was prepared and used as sorbent at ambient conditions for the microextraction and preconcentration of three different categories of pharmaceutical products as CLOM, KET and LOP in wastewater and human urine samples (Scheme 3). Hydrophobic moiety interaction with the long chain of the MIL as well as dipole–dipole between the OH and carbonyl group of the LOP with NH3+ cationic center of the MIL at neutral pH can be attributed to the successful extraction of the drug. Also, deprotonation of the OH− group of the KETO promoted electrostatic as well as hydrophobic interaction between the MIL and the analyte. CLOM is also effectively extracted through hydrophobic interaction by the long carbon chains of the sorbent and the π–π interaction due to the presence of benzene rings of the drug at neutral pH.

Shows structures of a KETO b LOP c CLOM structures
Important factors affecting the D-µSPE like elution solvent, pH, elution solvent volume, desorption time, sorbent type and dosage, extraction time, and salt concentration were optimized. It is observed that [OA]FeCl4 sorbent can successfully be used for the extraction of the three categories of drugs from both wastewater and human urine samples.
3.1 Characterization of the MIL Sorbent and the Intermediate Compound
Characterization of MILs is fundamental, because their physical properties can easily be affected by impurities and changes in structure [45, 46]. Therefore, several analytical methods are used to establish both their chemical and physical properties. Although it is difficult to quantify the impurities level, the characterizations aid in confirming the structure of the MILs. One such good example is when MIL is contaminated with lithium chloride, because both ions will not appear in standard mass spectrometry although their trace contamination will have no major influence on the elemental analysis. The most common contamination or impurities in MILs can be categorized into the water, unreacted starting material, solvent remains, metals, and halides [47]. In this regard, some of the characterization techniques used in this study can be summarized as Fourier-transform infrared spectroscopy (FTIR), zeta potential, magnetic property measurement systems (MPMS), proton nuclear magnetic resonance (1HNMR), thermogravimetric analysis (TGA), scanning electron microscopy, energy-dispersive X-ray analysis (SEM EDX) and Raman scattering spectroscopy.
3.1.1 1H NMR Spectra
The 1H‐NMR spectra were obtained on a JEOL spectrometer (JNM‐LA 500 MHz, JEOL USA Inc.). The 1H NMR of both the starting material (oleylamine) (Fig. 1) and the quaternary intermediate [OA]Cl (Fig. 2) were performed to investigate the protonation of the amine group in the parent compound after the addition of HCl. The 1H NMR spectra showed a downfield shift in the proton chemical shift from 4.81 ppm to 5.40 ppm. This shift is due to the hydrogen bonding between the amine group of the oleylamine and the hydrogen ion of the HCl acid. It can also be deduced that the electronic structure of the long hydrophobic chain cation forms acidic carbons at the consequent carbon numbers. No extra peaks were observed, thus indicating the purity of the intermediate. The peak observed at around 7.2 is because of the standard deuterated chloroform (CDCl3) solvent.

1HNMR of oleylamine

1HNMR of intermediate [OA]Cl
3.1.2 FT-IR Spectra
To understand the functional groups of the MIL sorbent, FT-IR was performed between 500 cm−1 and 4000 cm−1 using instrument Smart iTR NICOLET iS10 model from ThermoFisher scientific. The FTIR of parent compound oleylamine, the intermediate [OA]Cl, and the final product [OA]FeCl were compared. Oleylamine shows sharp dominant peaks at around 2917.7 cm−1 and around 2800 cm−1 which is due to methyl asymmetric stretching of CH3 and CH2, respectively (Fig. 3a). A peak at 1614.1 cm−1 is ascribed to the combined motions from NH2 scissoring and N–H bending. The N–H wagging is seen at around 750 cm−1 and the N–H bending mode is observed at 900 cm−1. Also, N–H stretching vibrations were observed at 3392.2 cm−1. Similar peaks were observed in the FTIR spectrum of the intermediate (Fig. 3b), although slight broadening was noted which can be attributed to the addition of the proton. The final complex FTIR spectrum (Fig. 3c) shows broad band stretching from 3500 cm−1 to 3300 cm−1 (circled) eclipsing the NH3 stretching region which can be associated with hygroscopic behavior of FeCl4− bonding.

FT-IR spectra of a oleylamine b intermediate [OA]Cl c MIL [OA]FeCl4
3.1.3 Raman Spectroscopy
The Raman spectroscope model LabRam instrument was used to record the Raman scattering of the MIL functional groups from 100 to 1000 cm−1 with laser excitation wavelength of 633 nm. A dominant sharp broad peak is seen at 332.4 nm (Fig. 4) which is attributed to symmetric bond stretching vibrations coming from Fe-Cl of FeCl4− as reported elsewhere [45, 48,49,50].

Raman spectra of [OA]FeCl4
3.1.4 Scanning Electron Microscopy (SEM)
The morphology of the material was also investigated and recorded using the JEOL JSM-6610LV model. The images (Fig. 5a and b), captured at micro-levels, indicated bright images due to the presence of highly conductive FeCl4− which largely appears on the surface of the MIL. Besides, the EDX elemental analysis confirmed the formation of the compound in the complex. No agglomeration was observed in the micrograph; however, the particles appear to have irregular shapes and sizes.

SEM image of a 10 μm b 5 μm, c and d respective elemental analysis in EDX images of [OA]FeCl4
3.1.5 Thermogravimetric (TGA) Analysis
Thermogravimetric (TGA) analysis of the MIL was conducted using the thermogravimetric analyzer model SDT Q600 instrument. First, around 8.5 mg of the MIL alumina pan is heated at high temperature up to 900 °C at the rate of 10 °C per min under nitrogen flow, and the decomposition pattern is recorded. As shown in Fig. 6a, there was no substantial weight loss until at around 243.3 °C, and at 500 °C, the weight loss was around 40%. The peak observed at around 58 °C is arising from the endothermic behavior of the MIL due to the melting point. In general decomposition of the material begins by a nucleophilic attack from the anion part of the MIL to the cation part [51]. This finding ascertains that the material has excellent thermal stability even at high temperatures.

a TGA analysis curve of [OA]FeCl4; b Zeta potential vs. pH of the [OA]FeCl4; c Magnetization of [OA]FeCl4 as a function of temperature under applied magnetic field at 5000–80,000 Oe and d Magnetization as a function of the applied magnetic field at 3–300 K temperature
3.1.6 Zeta Potential
The zeta potential was evaluated using the Zeta Potential Analyzer Zeta PALS model instrument. Six pH values 2, 4, 6, 8, 10, and 12 were examined by dispersing 10 mg of the MIL in 10 mL of double distilled water through sonication for 1 h. The Zeta potential is an important parameter to explain the stability of the particles. It is observed that the material has a larger zeta potential value of 15.9 mV indicating that the material is stabilized in electrostatic repulsion [52]. The MIL displays a positive charge at low pH and a negative charge at a higher pH with an isoelectric point at around 3.5 (Fig. 6b). The positive charge can be attributed to the cationic part of the MIL, while the negative charge can be due to the presence of the anionic part FeCl4−.
3.1.7 Magnetic Property Analysis
Magnetic property measurements were performed using the Physical Property Measurement System (PPMS), DaynaCool (Quantum Design, US) instrument. Approximately, 24.1 mg of the MIL sample was loaded to measure the magnetic susceptibility over a temperature range (0–300 K). Equally the magnetic field was varied from 5000–80,000 Oe. The magnetic susceptibility was found to decrease as the temperature increases from 0–300 K over all the magnetic fields studied (Fig. 6c). Besides, temperature dependency on the magnetization has been evaluated from 3–300 K and the relationship between temperature and magnetic moment is in perfect agreement with Curie–Weiss law (Fig. 6d). The result indicates a similar trend and characteristics reported in published literature for iron-containing magnetic ionic liquids for all the tested temperatures [45, 53]. In addition, a linear relationship between the magnetization of the MIL and the applied magnetic field was obtained at 300 K (room temperature) (Fig. 6c). The slope of the fitted line (red) shows that the magnetic susceptibility was found to be 5.89 which is much higher than the reported MILs as shown in Table 1. This direct relationship between magnetic moment and magnetic field implies that the synthesized MIL is paramagnetic and that there is no strong coupling arising from cations spin angular momenta.
3.2 Optimization of the Extraction Conditions
Various factors affecting extraction, such as elution solvent, pH, elution solvent volume, desorption time, sorbent dosage, extraction time, and salt concentration, were optimized to investigate the reliability of the developed method. Triplets were performed in all the experiments at ambient conditions.
3.2.1 Effect of Sorbent Amount
The effect of sorbent dosage was studied by varying the amount of sorbent from 10–50 mg. The sorbents were dispersed in the sample solution and all other parameters were kept constant. It was noted that as the dosage increases the extraction efficiency is enhanced up to 40 mg. Further sorbent addition did not significantly improve the extraction efficiency. This is generally expected since the addition of more sorbent results to more available active sites for the adsorption of the analytes. However, addition of the MIL sorbent beyond 40 mg appears to saturate the system, thus resulting in maximum extraction. Therefore, 40 mg was considered the best dosage for the extraction of the three analytes where equilibrium was obtained showing excellent extraction efficiency.
3.2.2 Effect of Sample pH
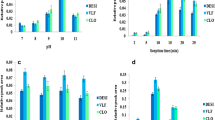
The pH value is one of the important parameters in extraction, because it influences analytes form, changes charge type, and as well as the density of the sorbent surface [57]. To evaluate the effect of the pH, the sample solution was adjusted from 2 to 9 and it was observed that neutral pH is the optimum for all the analytes under this study (Fig. 7a). In addition, pKa values of CLOM, LOP, and KETO were reported to be 9.4, 8.5, and 4.4, respectively, which is still above the isoelectric point [58,59,60]. The drugs are effectively extracted through hydrophobic interaction by the long carbon chains of the sorbent and the presence of benzene rings of the analytes at neutral pH. Generally, at lower pH, an electrostatic repulsion occurs between the positive charge of the analyte and the cationic part of the MIL, and by increasing the pH above 7 the efficiency reduces due to decreased cationic interaction of π-bonding. However, majorly hydrophobic interaction between the MIL and the analytes is responsible for the successful extraction of the compounds. Therefore, neutral pH 7 is considered to be the most suitable for the effective preconcentration of the analytes, and performance was noted to reduce as the pH rises to 9 in all the analytes and this is due to reduced molecular interaction as most of the analytes exist in their ionic form in the aqueous media.

Effect of a sample pH, b NaCl concentration, c extraction time and d desorption time on the extraction performance of the drugs on [OA]FeCl4 sorbent. All optimizations were performed in a 10 mL deionized water sample solution, spiked with 300 μg/L of the analyte
3.2.3 Effect of Salt Concentration
The salt concentration of the sample solution was evaluated using sodium chloride salt and the recovery rate was observed at different salt concentrations as 0%, 5%, 10%, 20%, and 30% w/v (Fig. 7b). The introduction of salt into the sample solution induces ionic strength hence reduced solubility of the drugs which enhanced their extraction. In this study, it is observed that as salt concentration increases, recovery rate is improved; however best condition was achieved at 20% w/v, and further studies were performed with 20% w/v.
3.2.4 Effect of Extraction and Desorption Time
Extraction time influences both analysis speed and extraction performance. To obtain better sensitivity, high speed, and precision, it is vital to identify the best extraction time that can give the right equilibrium between the sorbent and the sample solution. Therefore, the time taken by the sorbent to completely adsorb the analytes is crucial in the extraction process. To understand the efficient contact time, different extraction times were performed ranging from 5 to 60 min (Fig. 7c). It is observed that maximum analytical signal was achieved in 45 min and that can be associated with the large surface area of the sorbent and efficient contact between the sorbent and the analytes in the solution. Besides, the time taken to elute the analytes from sorbent was investigated in the same time range in ultrasonication and it was found that equally 45 min was enough to completely desorb the analytes from the sorbent (Fig. 7d). The negligible decrease recorded with increase in extraction time to 60 min can be attributed to solvent evaporation and back-extraction of the compounds and a similar trend has been reported when ketoprofen and other NSAIDS were extracted in water samples [61].
3.2.5 Desorption Solvent and its Volume
Micelles structured sorbents have been reported to be easily disturbed by organic solvents causing the desorption of the analytes after extraction. Therefore, in this study, we have examined the desorption of the analytes in several water-miscible solvents, such as ethanol, methanol, acetone, and acetonitrile. It was observed that acetonitrile showed the maximum recovery for almost all the analytes. However, LOP showed slightly better recovery in ethanol and methanol but with a negligible difference when compared with acetonitrile (Fig. 8(a)). Based on these findings, acetonitrile was chosen as the best desorption solvent and a similar finding was reported in the literature [62,63,64]. Subsequently, the optimum acetonitrile desorption volume was investigated by varying it from 100 to 500 μL. The finding indicated that 400 μL was sufficient to completely recover and elute the analytes from the sorbent (Fig. 8b). Hence, 400 μL of acetonitrile was employed as eluent for the rest of the study.

Effect of a solvent type, and b desorption volume. Optimization was performed in a 10 mL deionized water sample solution, spiked with 300 μg/L of the analyte
3.3 Figures of Merit of MIL-D-μ-SPE Method
The analytical performance of the developed extraction method was tested by using standard solutions of the three analytes under the optimized conditions (Table 2). The calibration curve of the analytes was obtained in a range of concentrations from 1 to 750 μg/L in 10 mL sample solution in double-distilled water. Regression coefficient (r2) was found to be 0.9974, 0.9967, and 0.9931 for CLOM, KETO, and LOP, respectively. The limit of detection (LOD) was calculated as three times the ratio of the standard deviation of the signal from the blank divided by the slope of the calibration curve and was found to be 2.3, 2.4, and 2.9 μg/L for CLOM, KETO, and LOP, respectively. The relative standard deviation (RSD, %) was calculated to be 3.1, 2.6 and 3.0 (n = 3) for inter-day and 2.7, 3.0 and 3.7 (n = 3) for intra-day for CLOM, KETO and LOP, respectively. The enrichment (EF) was defined as the ratio of the sample volume (10 mL) over the final desorption volume (400 µL) which is 25 for all the analytes. The lowest in the linear dynamic range was selected as the LOQ value (1 μg/L) for all.
3.4 Real Samples Analysis
To investigate the applicability and accuracy of the proposed method, complex samples of human urine and wastewater samples were performed. The samples were first spiked with analytes concentrations of 1, 250, and 750 μg/L under the optimized conditions and the results are shown in Table 3 with 3 replicate analyses of the complex samples, indicating perfect agreement with the spiked amount. Furthermore, Fig. 9 is a comparison of the chromatogram of 250 μg/L spiked and unspiked wastewater and human urine samples. The results obtained show similar values to the ones found when double-distilled was used. The samples were also not contaminated as no new peaks were detected in the analytes’ region with the unspiked samples.

HPLC–DAD chromatograms 250 μg/L of urine and wastewater samples after optimization. Peaks at 4.7 min, 5.9 min, and 9.5 min are corresponding to CLOM, LOP, and KETO, respectively
3.5 Comparison of the Method with Published Literature
The analytical method is compared with other reported published work for the preconcentration and determination of CLOM, LOP, and KETO as shown in Table 4. The method exhibits low detection for the simultaneous preconcentration of the analytes showing high sensitivity which is comparable or even better to the reported chromatographic methods. Furthermore, the method is cost-effective in the fact that three different categories of drugs are preconcentrated simultaneous in less than 10 min of retention time. In addition, the method has a much better RSD % than almost all the reported work with comparable LODs and linearity. The other important aspect is excellent high linearity range of up to 750 μg/L which is applicable to two complex matrixes wastewater and human urine unlike other method which largely concentrated on only one matrix, especially water.
3.6 Reuse and Recycle of the Sorbent
The reusability of the MIL was also studied. No physical destruction nor substantial loss of the sorbent was recorded; however, a negligible amount was lost during centrifugation and washing. First, the sorbent is washed thoroughly with double distilled water and checked for any residues of the analytes before reuse. The sorbent is oven-dried overnight at around 45 °C before the next extraction application. The material was tested at least three consecutive cycles and it was found that the extraction efficiency has not changed significantly with still more than 93% performance. The reusability of the MIL makes the extraction method more appealing and eco-friendly.
4 Conclusions
A complete microextraction, recovery and detection of trace pharmaceutical products from complex matrixes is vital in both biological and wastewater samples. Therefore, in this study, MIL [OA]FeCl4 was successfully synthesized and characterized as a sorbent for D-µSPE and quantification of three different categories of drugs clomipramine (CLOM), ketoprofen (KETO), and loperamide (LOP) in wastewater and urine samples as a model in an analytical problem. To understand the chemical and physical properties including the morphology several characterization techniques have been performed. The proposed method has the advantage of ease preparation of the sorbent, simultaneous extraction, and quantification of three different classes of drugs, cost-effective as well as reliable efficiency in recovery with decreased organic solvent consumption. The performance can be attributed to the hydrophobicity and electrostatic interaction of both the analytes and the sorbent enhancing ease extraction in the sample solution and desorption in the organic solvent. This proposed method showed better and comparable results with the existing ones, such as linearity of 1–750 μg/L, LOD values of 2.3—2.9 μg/L and RSD of 2.7—3.7%. The proposed method demonstrates a new direction in research where simple MILs can be used as efficient sorbent in µSPE without any modification for the simultaneous preconcentration of different types of drugs. Besides the method is eco-friendly as the sorbent can be reused several times. In this regard MIL [OA]FeCl4 can be used as an excellent alternative sorbent for simultaneous dispersive microextraction of CLOM, KETO and LOP drugs from wastewater effluents.
References
Daryanavard, S.M.; Zolfaghari, H.; Abdel-Rehim, A.; Abdel-Rehim, M.: Recent applications of microextraction sample preparation techniques in biological samples analysis. Biomed. Chromatogr. (2021). https://doi.org/10.1002/bmc.5105
Mansour, F.R.; Danielson, N.D.: Separation methods for captopril in pharmaceuticals and biological fluids. J. Sep. Sci. 35, 1213–1226 (2012)
Montesdeoca-Esponda, S.; Mahugo-Santana, C.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J.: A dispersive liquid–liquid micellar microextraction for the determination of pharmaceutical compounds in wastewaters using ultra-high-performace liquid chromatography with DAD detection. Biomed. Chromatogr. 29, 353–356 (2015)
Rezaee, M.; Assadi, Y.; Milani Hosseini, M.R.; Aghaee, E.; Ahmadi, F.; Berijani, S.: Determination of organic compounds in water using dispersive liquid-liquid microextraction. J. Chromatogr. A. 1116, 1–9 (2006). https://doi.org/10.1016/j.chroma.2006.03.007
Mansour, F.R.; Khairy, M.A.: Pharmaceutical and biomedical applications of dispersive liquid–liquid microextraction. J. Chromatogr. B. 1061, 382–391 (2017)
Rykowska, I.; Ziemblińska, J.; Nowak, I.: Modern approaches in dispersive liquid-liquid microextraction (DLLME) based on ionic liquids: A review. J. Mol. Liq. 259, 319–339 (2018)
Guo, L.; Lee, H.K.: Vortex-assisted micro-solid-phase extraction followed by low-density solvent based dispersive liquid–liquid microextraction for the fast and efficient determination of phthalate esters in river water samples. J. Chromatogr. A. 1300, 24–30 (2013). https://doi.org/10.1016/j.chroma.2013.01.030
Sajid, M.: Porous membrane protected micro-solid-phase extraction: A review of features, advancements and applications. Anal. Chim. Acta. 965, 36–53 (2017). https://doi.org/10.1016/j.aca.2017.02.023
Tran-Lam, T.-T.; Hong, M.B.T.; Le, G.T.; Luu, P.D.: Auramine O in foods and spices determined by an UPLC-MS/MS method. Food Addit. Contam. Part B. 13, 171–176 (2020)
Li, J.; Liu, J.; Lu, W.; Gao, F.; Wang, L.; Ma, J.; Liu, H.; Liao, C.; Chen, L.: Speciation analysis of mercury by dispersive solid-phase extraction coupled with capillary electrophoresis. Electrophoresis 39, 1763–1770 (2018)
Kepekci-Tekkeli, S.E.; Durmus, Z.: Magnetic solid phase extraction applications combined with analytical methods for determination of drugs in different matrices review. J. Chil. Chem. Soc. 64, 4448–4458 (2019)
Bello-López, M.Á.; Ramos-Payán, M.; Ocaña-González, J.A.; Fernández-Torres, R.; Callejón-Mochón, M.: Analytical applications of hollow fiber liquid phase microextraction (HF-LPME): a review. Anal. Lett. 45, 804–830 (2012)
Kataoka, H.: Recent advances in solid-phase microextraction and related techniques for pharmaceutical and biomedical analysis. Curr. Pharm. Anal. 1, 65–84 (2005)
Kataoka, H.: Current developments and future trends in solid-phase microextraction techniques for pharmaceutical and biomedical analyses. Anal. Sci. 27, 893 (2011)
Lasarte-Aragonés, G.; Lucena, R.; Cárdenas, S.: Effervescence-assisted microextraction—one decade of developments. Molecules 25, 6053 (2020)
Tanimu, A.; Jillani, S.M.S.; Alluhaidan, A.A.; Ganiyu, S.A.; Alhooshani, K.: 4-phenyl-1,2,3-triazole functionalized mesoporous silica SBA-15 as sorbent in an efficient stir bar-supported micro-solid-phase extraction strategy for highly to moderately polar phenols. Talanta 194, 377–384 (2019). https://doi.org/10.1016/j.talanta.2018.10.012
Tanimu, A.; Alhooshani, K.: N-sulfonyl-4-hydroxymethyl-1,2,3-triazole functionalized SBA-15: A porous organic-inorganic material for trace-level phenolic compounds extraction from water samples by stir bar-supported micro-solid-phase extraction. Microchem. J. 159, 105410 (2020). https://doi.org/10.1016/j.microc.2020.105410
Banitaba, M.H.; Davarani, S.S.H.; Ahmar, H.; Movahed, S.K.: Application of a new fiber coating based on electrochemically reduced graphene oxide for the cold-fiber headspace solid-phase microextraction of tricyclic antidepressants. J. Sep. Sci. 37, 1162–1169 (2014). https://doi.org/10.1002/jssc.201301369
Arabi, M.; Ostovan, A.; Bagheri, A.R.; Guo, X.; Wang, L.; Li, J.; Wang, X.; Li, B.; Chen, L.: Strategies of molecular imprinting-based solid-phase extraction prior to chromatographic analysis. TrAC Trends Anal. Chem. 128, 115923 (2020)
Lu, W.; Ming, W.; Zhang, X.; Chen, L.: Molecularly imprinted polymers for dispersive solid-phase extraction of phenolic compounds in aqueous samples coupled with capillary electrophoresis. Electrophoresis 37, 2487–2495 (2016)
Qin, D.; Wang, J.; Ge, C.; Lian, Z.: Fast extraction of chloramphenicol from marine sediments by using magnetic molecularly imprinted nanoparticles. Microchim. Acta. 186, 1–10 (2019)
Lu, W.; Liu, J.; Li, J.; Wang, X.; Lv, M.; Cui, R.; Chen, L.: Dual-template molecularly imprinted polymers for dispersive solid-phase extraction of fluoroquinolones in water samples coupled with high performance liquid chromatography. Analyst. 144, 1292–1302 (2019)
Chisvert, A.; Cárdenas, S.; Lucena, R.: Dispersive micro-solid phase extraction. TrAC - Trends Anal. Chem. 112, 226–233 (2019). https://doi.org/10.1016/j.trac.2018.12.005
Awual, M.R.; Yaita, T.; Kobayashi, T.; Shiwaku, H.; Suzuki, S.: Improving cesium removal to clean-up the contaminated water using modified conjugate material. J. Environ. Chem. Eng. 8, 103684 (2020)
Awual, M.R.: A novel facial composite adsorbent for enhanced copper (II) detection and removal from wastewater. Chem. Eng. J. 266, 368–375 (2015)
Kamel, R.M.; Shahat, A.; Hegazy, W.H.; Khodier, E.M.; Awual, M.R.: Efficient toxic nitrite monitoring and removal from aqueous media with ligand based conjugate materials. J. Mol. Liq. 285, 20–26 (2019)
Awual, M.R.; Hasan, M.M.: A ligand based innovative composite material for selective lead (II) capturing from wastewater. J. Mol. Liq. 294, 111679 (2019)
Khandaker, S.; Toyohara, Y.; Saha, G.C.; Awual, M.R.; Kuba, T.: Development of synthetic zeolites from bio-slag for cesium adsorption: Kinetic, isotherm and thermodynamic studies. J. Water Process Eng. 33, 101055 (2020)
Awual, M.R.; Hasan, M.M.; Iqbal, J.; Islam, M.A.; Islam, A.; Khandaker, S.; Asiri, A.M.; Rahman, M.M.: Ligand based sustainable composite material for sensitive nickel (II) capturing in aqueous media. J. Environ. Chem. Eng. 8, 103591 (2020)
Islam, A.; Ahmed, T.; Awual, M.R.; Rahman, A.; Sultana, M.; Abd Aziz, A.; Monir, M.U.; Teo, S.H.; Hasan, M.: Advances in sustainable approaches to recover metals from e-waste-A review. J. Clean. Prod. 244, 118815 (2020)
Awual, M.R.; Hasan, M.M.; Iqbal, J.; Islam, A.; Islam, M.A.; Asiri, A.M.; Rahman, M.M.: Naked-eye lead (II) capturing from contaminated water using innovative large-pore facial composite materials. Microchem. J. 154, 104585 (2020)
Shahat, A.; Hassan, H.M.A.; El-Shahat, M.F.; El Shahawy, O.; Awual, M.R.: Visual nickel (II) ions treatment in petroleum samples using a mesoporous composite adsorbent. Chem. Eng. J. 334, 957–967 (2018)
Awual, M.R.; Hasan, M.M.; Islam, A.; Rahman, M.M.; Asiri, A.M.; Khaleque, M.A.; Sheikh, M.C.: Introducing an amine functionalized novel conjugate material for toxic nitrite detection and adsorption from wastewater. J. Clean. Prod. 228, 778–785 (2019)
Toledo-Neira, C.; Álvarez-Lueje, A.: Ionic liquids for improving the extraction of NSAIDs in water samples using dispersive liquid–liquid microextraction by high performance liquid chromatography-diode array–fluorescence detection. Talanta 134, 619–626 (2015). https://doi.org/10.1016/j.talanta.2014.11.067
Sajid, M.: Trends in analytical chemistry magnetic ionic liquids in analytical sample preparation : A literature review. Trends Anal. Chem. 113, 210–223 (2019). https://doi.org/10.1016/j.trac.2019.02.007
Hassan, A.A.; Sajid, M.; Al Ghafly, H.; Alhooshani, K.: Ionic liquid-based membrane-protected micro-solid-phase extraction of organochlorine pesticides in environmental water samples. Microchem. J. 158, 105295 (2020)
Hassan, A.A.; Sajid, M.; Tanimu, A.; Abdulazeez, I.; Alhooshani, K.: Removal of methylene blue and rose bengal dyes from aqueous solutions using 1-naphthylammonium tetrachloroferrate (III). J. Mol. Liq. (2020). https://doi.org/10.1016/j.molliq.2020.114966
Ge, D.; Lee, H.K.: Ionic liquid based dispersive liquid–liquid microextraction coupled with micro-solid phase extraction of antidepressant drugs from environmental water samples. J. Chromatogr. A. 1317, 217–222 (2013). https://doi.org/10.1016/j.chroma.2013.04.014
Alves, V.; Gonçalves, J.; Conceição, C.; Teixeira, H.M.; Câmara, J.S.: An improved analytical strategy combining microextraction by packed sorbent combined with ultra high pressure liquid chromatography for the determination of fluoxetine, clomipramine and their active metabolites in human urine. J. Chromatogr. A. 1408, 30–40 (2015). https://doi.org/10.1016/j.chroma.2015.07.021
Miller, H.; Panahi, L.; Tapia, D.; Tran, A.; Bowman, J.D.: Loperamide misuse and abuse. J. Am. Pharm. Assoc. 57, S45–S50 (2017)
Li, W.; Wang, R.; Chen, Z.: Zr-based metal-organic framework-modified cotton for solid phase micro-extraction of non-steroidal anti-inflammatory drugs. J. Chromatogr. A. 1576, 19–25 (2018). https://doi.org/10.1016/j.chroma.2018.09.032
Seidi, S.; Rezazadeh, M.; Yamini, Y.: Pharmaceutical applications of liquid-phase microextraction. TrAC Trends Anal. Chem. 108, 296–305 (2018). https://doi.org/10.1016/j.trac.2018.09.014
Siddiqui, M.R.; AlOthman, Z.A.; Rahman, N.: Analytical techniques in pharmaceutical analysis: A review. Arab. J. Chem. 10, S1409–S1421 (2017). https://doi.org/10.1016/j.arabjc.2013.04.016
Hassan, A.A.; Tanimu, A.; Alhooshani, K.: Iron and cobalt-containing magnetic ionic liquids for dispersive micro-solid phase extraction coupled with HPLC-DAD for the preconcentration and quantification of carbamazepine drug in urine and environmental water samples. J. Mol. Liq. 336, 116370 (2021). https://doi.org/10.1016/j.molliq.2021.116370
Wang, J.; Yao, H.; Nie, Y.; Zhang, X.; Li, J.: Synthesis and characterization of the iron-containing magnetic ionic liquids. J. Mol. Liq. 169, 152–155 (2012). https://doi.org/10.1016/j.molliq.2012.02.005
Seddon, K.R.; Stark, A.; Torres, M.-J.: Influence of chloride, water, and organic solvents on the physical properties of ionic liquids. Pure Appl. Chem. 72, 2275–2287 (2000)
Clare, B., Sirwardana, A., MacFarlane, D.R.: Synthesis, purification and characterization of ionic liquids. Ion. Liq. 1–40 (2009)
Jiang, W.; Zhu, W.; Li, H.; Xiong, J.; Xun, S.; Zhao, Z.; Wang, Q.: Deep oxidative desulfurization of fuels catalyzed by magnetic Fenton-like hybrid catalysts in ionic liquids. RSC Adv. 3, 2355–2361 (2013). https://doi.org/10.1039/C2RA22227B
Ullah, S.; Bustam, M.A.; Al-Sehemi, A.G.; Assiri, M.A.; Gonfa, G.; Mukhtar, A.; Ayoub, M.; Ahmad, T.: Experimental investigations on the regeneration of desulfurized 1-butyl-3-methylimidazolium tetrachloroferrate [Bmim][FeCl4] and 1-butyl-3-methylimidazolium thiocyanate [Bmim][SCN] ionic liquids: A raman spectroscopic study. J. Raman Spectrosc. 51, 546–554 (2020). https://doi.org/10.1002/jrs.5784
Papatheodorou, G.N., Kalampounias, A.G., Yannopoulos, S.N.: Raman Spectroscopy of High Temperature Melts in Molten Salts and Ionic Liquids: Never the Twain? Molten Salts Ion. Liq. 301–340 (2012)
Clough, M.T.; Geyer, K.; Hunt, P.A.; Mertes, J.; Welton, T.: Thermal decomposition of carboxylate ionic liquids: trends and mechanisms. Phys. Chem. Chem. Phys. 15, 20480–20495 (2013)
Dong, B.; Su, Y.; Liu, Y.; Yuan, J.; Xu, J.; Zheng, L.: Dispersion of carbon nanotubes by carbazole-tailed amphiphilic imidazolium ionic liquids in aqueous solutions. J. Colloid Interface Sci. 356, 190–195 (2011)
Santos, E.; Albo, J.; Irabien, A.: Magnetic ionic liquids: synthesis, properties and applications. Rsc Adv. 4, 40008–40018 (2014)
Santos, E.; Albo, J.; Rosatella, A.; Afonso, C.A.M.; Irabien, Á.: Synthesis and characterization of Magnetic Ionic Liquids (MILs) for CO2 separation. J. Chem. Technol. Biotechnol. 89, 866–871 (2014). https://doi.org/10.1002/jctb.4323
Hayashi, S.; Saha, S.; Hamaguchi, H.: A new class of magnetic fluids: bmim [fecl/sub 4/] and nbmim [fecl/sub 4/] ionic liquids. Ieee Trans. Magn. 42, 12–14 (2005)
Del Sesto, R.E., McCleskey, T.M., Burrell, A.K., Baker, G.A., Thompson, J.D., Scott, B.L., Wilkes, J.S., Williams, P.: Structure and magnetic behavior of transition metal based ionic liquids. Chem. Commun. 447–449 (2008)
Kazemi, E.; Shabani, A.M.H.; Dadfarnia, S.; Abbasi, A.; Vaziri, M.R.R.; Behjat, A.: Development of a novel mixed hemimicelles dispersive micro solid phase extraction using 1-hexadecyl-3-methylimidazolium bromide coated magnetic graphene for the separation and preconcentration of fluoxetine in different matrices before its determination b. Anal. Chim. Acta. 905, 85–92 (2016)
Petersen, N.J.; Jensen, H.; Hansen, S.H.; Foss, S.T.; Snakenborg, D.; Pedersen-Bjergaard, S.: On-chip electro membrane extraction. Microfluid. Nanofluidics. 9, 881–888 (2010)
Zgoła-Grześkowiak, A.: Application of DLLME to isolation and concentration of non-steroidal anti-inflammatory drugs in environmental water samples. Chromatographia 72, 671–678 (2010)
Huang, F.; Qu, S.; Zhang, S.; Liu, B.; Kong, J.: Sensitive voltammetric detection of clomipramine at 16-mercapto-hexadecanoic acid self-assembled monolayer modified gold electrode. Microchim. Acta. 161, 149–155 (2008)
Abd Wahib, S.M.; Wan Ibrahim, W.A.; Sanagi, M.M.; Kamboh, M.A.; Abdul Keyon, A.S.: Magnetic sporopollenin-cyanopropyltriethoxysilane-dispersive micro-solid phase extraction coupled with high performance liquid chromatography for the determination of selected non-steroidal anti-inflammatory drugs in water samples. J. Chromatogr. A. 1532, 50–57 (2018). https://doi.org/10.1016/j.chroma.2017.11.059
Chaves, A.R.; Silva, S.M.; Queiroz, R.H.C.; Lanças, F.M.; Queiroz, M.E.C.: Stir bar sorptive extraction and liquid chromatography with UV detection for determination of antidepressants in plasma samples. J. Chromatogr. B. 850, 295–302 (2007). https://doi.org/10.1016/j.jchromb.2006.11.042
Kamaruzaman, S.; Sanagi, M.M.; Endud, S.; Ibrahim, W.A.W.; Yahaya, N.: MCM-41 solid phase membrane tip extraction combined with liquid chromatography for the determination of non-steroidal anti-inflammatory drugs in human urine. J. Chromatogr. B. 940, 59–65 (2013)
Klančar, A.; Zakotnik, M.; Roškar, R.; Trontelj, J.: Multi-residue analysis of selected pharmaceuticals in wastewater samples by stir-bar sorptive extraction followed by liquid desorption and liquid chromatography-mass spectrometry. Anal. Methods. 9, 5310–5321 (2017). https://doi.org/10.1039/C7AY01310H
Hamidi, F.; Hadjmohammadi, M.R.; Aghaie, A.B.G.: Ultrasound-assisted dispersive magnetic solid phase extraction based on amino-functionalized Fe3O4 adsorbent for recovery of clomipramine from human plasma and its determination by high performance liquid chromatography: Optimization by experimental desig. J. Chromatogr. B. 1063, 18–24 (2017)
Jalilian, N.; Asgharinezhad, A.A.; Ebrahimzadeh, H.; Molaei, K.; Karami, S.: Magnetic solid phase extraction based on modified magnetite nanoparticles coupled with dispersive liquid–liquid microextraction as an efficient method for simultaneous extraction of hydrophobic and hydrophilic drugs. Chromatographia 81, 1569–1578 (2018)
Di, X.; Zhao, X.; Guo, X.: Dispersive micro-solid phase extraction combined with switchable hydrophilicity solvent-based homogeneous liquid-liquid microextraction for enrichment of non-steroidal anti-inflammatory drugs in environmental water samples. J. Chromatogr. A. 1634, 461677 (2020)
Pantůčková, P.; Kubáň, P.: In-line coupling of supported liquid membrane extraction to capillary electrophoresis for simultaneous analysis of basic and acidic drugs in urine. J. Chromatogr. A. 1519, 137–144 (2017)
Acknowledgements
The financial support provided by the Deanship of Scientific Research (DSR), King Fahd University of Petroleum, through the grant No. DSR SL191003 is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hassan, A.A., Tanimu, A. & Alhooshani, K. Dispersive Micro-Solid Phase Extraction of Pharmaceutical Drugs from Wastewater and Human Urine Using (Z)-Octadec-9-en-1-aminium tetrachloroferrate (III) Ionic Liquid and Analysis by High-Performance Liquid Chromatography. Arab J Sci Eng 47, 6915–6929 (2022). https://doi.org/10.1007/s13369-021-06034-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13369-021-06034-3