
Abstract
The high surface area, lightweight and excellent biocompatibility of nanocellulose (NC) have resulted in its wide application as a bio-based reinforcement agent. In this study, raw oil palm fronds leaves (OPFL) were used as the cellulose source to prepare NC. Successive treatments of raw OPFL with bleach, alkali and acid yielded NC in a form of white powder. X-ray diffraction, transmission and field emission scanning electron micrographs showed that the isolated NC formed crystalline needlelike structures with diameters ranging between 10 and 30 nm. The crystallinity index of NC was 45.5%, greater than raw OPFL (25.3%) and cellulose (30.5%). The AFM micrographs showed that the NC exhibited a root-mean-square roughness of 9.146 nm. FTIR-ATR spectra revealed the disappearance of a C=O peak at 1732 cm−1 with the absence of impurities, while the EDX spectrum further affirmed that lignin and hemicellulose were successfully eliminated. BET surface area of NC (5.108 m2/g) was found higher than cellulose (1.4526 m2/g). The TGA data revealed that NC decomposed between 120 and 450 °C, with an 80% mass loss that corresponded to sulfate groups (O–SO3). In short, the nanometer dimensions of NC extracted from raw OPFL implied its promising applications as a reinforcing agent for improving mechanical properties of nanocomposites.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Oil palm (Elaeis guineensis) belongs to the family of Palmaceae which originated from West Africa: This crop was only introduced to the Southeast Asian region in the last century [1]. Oil palm is among the current important agronomic and economic bio-products of Malaysia [2] and will do so in the future, based on current rates of agricultural land expansion and oil palm replanting activities. However, the rapid growth of oil palm plantations has concomitantly left the country to deal with 50–80 million tons of biomass per hectare in 2012, in which the number is forecasted to exceed 100 million tons by 2020 [3]. In fact, oil palm leaves (OPFL) constitute an enormous 70% of biomass produced by the oil palm industry. This is a matter of concern as large quantities of biomass if not properly managed can potentially threaten the ecosystem [4]. This is because the covered parts of the plantation floor can be a breeding grown for various kinds of microorganisms that can potentially interfere with the ecological niche of an area. To get rid of the excess biomass, farmers in certain regions of the world often resort to the unpleasant practice of burning the OPFL. This conventional technique is quicker and fertilizes the land [5,6,7]. Hence, concerted efforts by the scientific community in search of novel uses for OPFL would facilitate effective removal and use of the biomass, and possibly, a source of revenue for the nation. In fact, the idea of using biomass as a source of polymer to prepare novel composites has grown substantially for the past few decades. This is considering their constant availability and renewability [6, 8,9,10].
Lignocellulosic fibers are useful as an alternative reinforcement agent in polymer matrices to prepare composites of different applications. Their relatively low densities and abrasiveness, high stiffness due to high possible filling levels, high specific properties and high bending resistance are the advantages associated with using natural fibers [11]. In fact, OPFL is reportedly an excellent source of renewable polymer to produce nanocellulose (NC) [12]. NC is valued for its nanoscale mechanical properties [13] that reinforce composites by forming a stable cross-linked network within the matrices [14]. Since matrix reinforcement by nanofillers from cellulosic material is largely contributed by the crystalline fraction of the nanomaterial, their incorporation can lead to favorable changes in properties of the resultant hybrid composite, thus widening its application. This is due to a percolation network that formed through multiple hydrogen bonds between the reactive surface hydroxyl side groups on NC with that of the polymer matrix, a crucial reinforcement feature when fabricating functional hybrid composites. Then again, the suitability of lignocellulosic-based reinforcement material for a given product or purpose also stems from other factors such as chemical, morphological and structural aspects, including the end use, and not the cellulose content, alone [11].
So far, techniques used to prepare NC from lignocellulosic materials generally require treatment using strong acids to disrupt the supramolecular structure of cellulose. The process expels the amorphous region in the fiber leaving behind only the crystalline parts [15, 16]. Moreover, a higher degree of crystallinity of the nanomaterial imparts higher mechanical properties and reduces flexibility and extensibility of the resultant composite. Khoo and his co-workers [17] successfully prepared NC from microcrystalline cellulose that showed dimensions between 250 and 300 nm in length and 10–20 nm in diameter using the acid hydrolysis process. Other techniques that used mechanical treatments to extract and prepare NC from biomass include ultrasonication and steam explosion. These techniques have been successfully used to produce NC from cellulose from pine wood [18], pineapple leaves [8] and jute fiber [19].
In the present study, NC was recovered from OPFL using a consecutive treatment of acid and base hydrolysis. Morphology of the nanomaterial was studied by Fourier transform infrared, field emission scanning electron microscopy, transmission electron microscopy and atomic force microscopy. The study also assessed the thermal properties of the treated OPFL fiber and NC using thermogravimetric analysis, while the crystallinity index of NC was estimated by X-ray diffraction.
2 Materials and Methods
2.1 Chemicals and Materials
Oil palm frond leaves (OPFL) were collected from an oil palm plantation within the grounds of Universiti Teknologi Malaysia (UTM). Sodium chlorite (NaClO2), glacial acetic acid, sodium hydroxide (NaOH) and sulfuric acid (H2SO4) were purchased from Sigma-Aldrich (St. Louis, USA). Chemicals used in this study were all analytical grade and used without further purification. Distilled water was prepared in our laboratory.
2.2 Production and Extraction of NC from OPFL
Raw OPFL were sorted, cleaned, cut and sun-dried for 3 days, before grinding using a blender and sieved into finer sizes. For the delignification process, buffer solution (acetic acid/sodium chlorite/distilled water, 0.06 mL:0.30 g:30 mL per 1 g of OPFL) was transferred into a 1-L round-bottom flask that contained OPFL powder (20 g), and the suspension was refluxed in a paraffin oil bath at 85 °C. At every 1-h interval, acetic acid and sodium chlorite were added to the mixture and this process was repeated five times. The mixture was left to stir at 70 °C for a further 15 h before rinsing with copious amounts of distilled water and centrifuged for 10 min at 8500 rpm. The process was repeated until a neutral pH was reached before air-drying in an oven at 70 °C overnight to obtain the “holocellulose” [20].
Next, the holocellulose was purified by alkali treatment to eliminate any remaining hemicellulose and lignin. Holocellulose was mixed with 17% (w/v) sodium hydroxide solution and left to stir for 5 h at room temperature. The suspension was rinsed in distilled water and centrifuged for 10 min at 8500 rpm, and the process was repeated until a neutral pH was reached. The resultant suspension was dried overnight in a 70 °C oven to afford the pure cellulose.
The process of NC extraction was conducted in Erlenmeyer flask to which sulfuric acid (10 mL, 9 M) was added to the cellulose, and the suspension was stirred at room temperature for 5 h. Then, distilled water was added to the mixture to stop the hydrolysis reaction. The suspension was rinsed with distilled water and centrifuged for 10 min at 8500 rpm, and the process was repeated till a neutral pH was reached. The resultant suspension was frozen at − 30 °C overnight and lyophilized to obtain the NC [20].
2.3 Characterization of Extracted NC
2.3.1 Fourier-Transform Infrared-Attenuated Total Reflection (FTIR-ATR)
Raw OPFL, cellulose and extracted NC were analyzed using FTIR-ATR. Spectra for all samples were collected using 32 scans per sample in the one-bounce ATR mode using a Spectrum 100 FTIR spectrometer (PerkinElmer, USA). A small amount of each sample was placed on the Diamond/ZnSe crystal plate, and the vibrational spectrum was recorded in transmission mode with a 4 cm−1 resolution for the spectral region between 400 and 4000 cm−1.
2.3.2 Thermogravimetric Analysis (TGA)
Thermogravimetric analysis was used to compare the thermal stabilities of different samples, to check for improved mechanical properties. Thermogravimetric analyses of the raw OPFL, cellulose and NC were conducted using a Thermogravimetric Analyzer 4000 (PerkinElmer). Samples (1–5 mg) were analyzed within a temperature range of 30–900 °C under N2 flow with a heating rate of 10 °C/min.
2.3.3 X-ray Diffraction (XRD)
Cellulose contained within the OPFL fibers may comprise amorphous and crystalline oriented zones in micro-or nanoscale, which imparts a certain degree of crystallinity. The percentage of crystallinity, to a certain extent, is proportional to the mechanical strength of a material [21]. X-ray diffractograms of the raw OPFL, cellulose and NC were recorded on a powder X-ray diffractometer (D/max 2200, Rigaku, Japan) equipped with Cu Kα radiation source (λ = 0.154 nm) operating at 40 kV and 30 mA. The XRD patterns were recorded over the angular range 2θ = 10–40° with the increment of 10° min−1. Crystallinity index (Ic) was estimated from the heights of the 002 peak (I200, 2θ = 22°) and the intensity minimum between the 002 and 101 peaks (Iam, 2θ = 15°) using the Segal method [22] (Eq. 1). Crystalline and amorphous material was represented by the term I002, whereas Iam denotes the amorphous material:
2.3.4 Nitrogen Adsorption
Surface area analysis of cellulose and NC was carried out using nitrogen adsorption (Micromeritics 3Flex 3.01: USA) instrument. The sample was first degassed at 35 °C under vacuum for 24 h. The sample tube was then placed on the analysis port, and the analysis was performed at 77 K. The specific surface areas of both samples were calculated from the linear region of the isotherms using the Brunauer–Emmett–Teller (BET) equation in a relative P/P0 pressure range of 0.0–0.6.
2.3.5 Field Emission Scanning Electron Microscopy (FESEM)
FESEM micrographs were taken using a JEOL JEM-6700F Field Emission Scanning Electron Microscopy (FESEM) equipped with an energy-dispersive X-ray analysis (EDX) operated with an acceleration voltage and electric current of 5 kV and 10 µA, respectively. A small amount of the sample was deposited on the surface of a silicon wafer followed by the sputter coating with a thin film of gold to avoid charging under the electron beam. The micrographs were collected between 500 and 15,000× magnification.
2.3.6 Transmission Electron Microscopy (TEM)
TEM micrographs were recorded on a Zeiss EFTEM Libra 120 Transmission Electron Microscope (TEM) equipped with an S-twin objective lens with a 2.0 nm or better point resolution for magnification between 10,000 and 20,000×, and it was mounted on a single-tilt holder with LCD camera. NC was dispersed in distilled water after ultrasonication and dropped on a carbon-coated copper grid, as reported in Rubentheren et al. [23]. The copper grid was stored in a dry cabinet for 48 h before viewing.
2.3.7 Atomic Force Microscopy (AFM)
Topographic information of the surface of NC was observed using the Atomic Force Microscope Seiko SPI-3800 N (Japan). The substrate surface was chemically treated with dimethyl sulfoxide (DMSO) and allowed to dry. NC powder was dusted over the entire surface of the DMSO-treated substrate to form a monolayer. AFM micrographs were then recorded in dynamic force mode, and the scans were performed on an area of 1.75–2.0 µm. The 3D profile of the nanomaterial was established by measuring forces between a sharp tip of AFM probe (less than 10 nm) and the surface of the composite at a very short distance (0.2–10 nm probe sample separation). Estimation of the root-mean-square (RMS) roughness of the sample was obtained from the topographical data of AFM and analyzed using a JPKSPM data processing software.
3 Results and Discussion
3.1 Extraction Protocol to Produce NC
Preparation of NC required the consecutive treatments of raw OPFL of bleaching, alkaline treatment and acid hydrolysis. The initial step of chlorite bleaching was crucial to extract “holocellulose” from the raw cellulose fibers in OPFL, in which most of the lignin contents responsible for defibrillation are eliminated [24]. The nucleophilic attack of the phenolic groups in lignin by the acidified sodium chlorite liberates chlorine dioxide [25] which then disintegrates the polymer material. However, the duration of the chlorite treatment on raw OPFL was limited to 15 h in this study, as it was time-wasting and led to an insignificant reduction in lignin content [24].
The subsequent step of alkaline pretreatment performed in this study was to solubilize the lignin and hemicellulose [26] remaining in the holocellulose (Fig. 1). It is an additional purification step in conjunction with other functions: (1) as a swelling agent; (2) to amplify the internal surface of the material; and (3) to reduce the polymerization rate and degree of crystallinity of cellulose, and lastly, (4) to break the glycosidic ether bonds that bind the lignin structure [27]. These changes synergistically intensify the susceptibility of cellulose in OPFL for transformation into NC as the lignin can no longer act as a protective shield [27]. Figure 1b depicts a pale beige-colored solid of purified cellulose fibers extracted from raw OPFL.

Photographs of a holocellulose, b cellulose and c NC
The purified cellulose was finally acid-hydrolyzed using sulfuric acid to cleave the glycosidic linkages in the supramolecular structure of cellulose. This converts the biopolymer into the nanoscale NC [12], leaving behind a white powder as shown in Fig. 1c. In this process, the amorphous regions of cellulose are eliminated, and only the crystalline portion remains, as previously described by Maiti et al. [15]. This invariably enhances the crystallinity and stability of the resultant NC. The final process introduces key anionic sulfate ester groups on the surface of NC, formed from the esterification of –OH groups on cellulose and–COOH groups in sulfuric acid [28]. Introduction of the negatively charged anionic sulfate ester groups on the surface of NC can avert the aggregation of minute NC structures via nucleophilic repulsions, thus facilitating the dispersion of NC [29].
3.2 Characterization of NC
3.2.1 Fourier-Transform Infrared-Attenuated Total Reflection (FTIR-ATR)
FTIR-ATR spectroscopy is a potent analytical tool for characterizing and elucidating structures of chemical compounds based on the characteristic frequencies at which their molecular functional groups vibrate [30]. In this study, the FTIR-ATR technique was performed to analyze the functional groups present in OPFL and to examine the changes that occurred prior to and after various chemical treatments. Spectra for the (a) raw OPFL, (b) cellulose and (c) NC are displayed in Fig. 2. Figure 2a for the untreated OPFL fiber reveals a peak at 1732 cm−1 that was associated with the C=O stretching of either uronic ester and acetyl groups in hemicellulose, or the carboxyl groups of the p-coumaric and ferulic acids of lignin [28, 31]. Vibrational stretching of C=C bonds in the benzene ring of lignin appeared as two peaks at 1630 cm−1 and 1521 cm−1, which was similar to a previous work on wheat straw biomass [32]. This was supported by characteristic peaks of lignin at 1431 cm−1 and 1232 cm−1, allotted to the methoxy O–CH3 and C–O–C stretching vibrations of the aryl–alkyl ether group, respectively [12, 33].

FTIR-ATR spectra of a OPFL, b cellulose and c NC
Absorption peaks found between regions 3288–3332 cm−1 and 2890–2918 cm−1 in all spectra were related to the stretching vibrations of O–H and aliphatic saturated C–H groups, respectively [12, 33]. The characteristic vibrational stretching observed in all three spectra for regions 1020–1032 cm−1 and 666–664 cm−1 corresponded to the stretching vibrations of both pyranose ring and glycosidic bonds between glucose units in cellulose [34]. The bending of O–H groups observed at 1633–1635 cm−1 was likely due to the adsorption of water molecules within the biopolymer network [35].
Spectra for cellulose and extracted NC in Fig. 2b, c depicted typical absorption peaks of expected functional groups in the raw OPFL (Fig. 2a). The absence of a C=O vibrational peak in the chemically treated OPFL fiber was the crucial differentiating factor that supported the elimination of hemicellulose after the alkali treatment. The emergence of two peaks at 1633 cm−1 and 1635 cm−1 in the treated OPFL fibers (Fig. 2b, c) was likely the result of NaOH that reacted with the O–H groups of cellulose to form water molecules within the cellulose fibers [36]. The study found that trapped water molecules within the cellulose molecules were tough to remove, despite the overnight drying process. This was directly due to the strong interaction between cellulose and water as previously described [19].
3.2.2 Thermogravimetric Analysis (TGA)
The thermal properties of raw OPFL, cellulose and NC were analyzed by TGA, and the results are presented in Fig. 3a, b. All thermograms showed minor mass losses between temperatures 50 and 150 °C that accompanied the evaporation of surface-adsorbed moisture [34, 35]. Thermal degradation of raw OPFL fiber occurred in two stages, as a result of different components decomposing under different temperatures [31]. The first mass loss observed at 170–370 °C (43.9%) ensued the thermal decomposition of hemicellulose and lignin, followed by a large mass loss between 370 and 600 °C (mass loss 16.5%) due to pyrolysis of cellulose. Studies by Deepa et al. [35] and Chieng et al. [3] also reported a similar thermal decomposition trend for sisal and oil palm mesocarp fiber, respectively.

a TGA and b DTG curves for the decomposition of raw OPFL, cellulose and NC
The thermogram for cellulose showed a dramatic decline in mass between 270 and 380 °C (mass loss 73.3%) related to the partial degradation of remaining hemicelluloses and lignin [35]. This was supported by the DTG profile that revealed the absence of a broad peak following the bleaching and alkali treatments of raw OPFL, indicative of the successful removal of hemicelluloses and lignin. The thermal stability of cellulose was seen also to improve over raw OPFL once the amorphous components were removed. Likewise, Moriana et al. [37] discovered that bleached materials and alkali branches of softwood forestry were thermally more stable than the respective raw materials since they were degraded within a narrower temperature range.
The T onset of NC produced after hydrolysis by sulfuric acid was lower than raw OPFL and cellulose. NC degradation that occurred between temperatures 120 and 450 °C (mass loss ~ 80%) implied the presence of sulfate groups (O–SO3) after the sulfonation of cellulose. According to the literature, the substitution of –OH groups by O–SO3 after acid hydrolysis produced NC that is easier to pyrolyze [38]. This explains the marginal reduction in the thermal stability of the nanomaterial. Furthermore, the high surface area of NC to a certain extent tends to expedite the heat transfer process and decomposition rate, as seen from the maximum thermal degradation temperature that was raised from 270 to 380 °C in cellulose to 120–450 °C in NC.
3.2.3 X-ray Diffraction (XRD)
The crystallinity of NC as estimated by XRD can be used to describe the changes in the physical and mechanical properties of the samples after a series of chemical treatments. Raw natural fibers are comprised of different elements which are either amorphous or crystalline [39], where hemicellulose and lignin belong to the amorphous region, whereas cellulose consists of both the amorphous and crystalline regions of the plant fiber [28]. In this study, the crystallinity index was used to compute the relative amounts of crystalline material/regions in the raw OPFL, cellulose and NC.
Figure 4 shows the X-ray patterns for (a) raw OPFL, (b) cellulose and (c) NC, respectively. As can be seen, the diffractograms for all samples revealed monoclinic sphenodic structures which are typical for cellulose-I polymorph, a ubiquitous form of cellulose [4]. The similar patterns between the three different samples strongly implied that pretreatment of raw OPFL did not significantly influence the natural cellulose-I structure. The marginally increased intensities of peaks at 2θ = 18° and 2θ = 21° were indicative of an increase in purity after the consecutive chemical pretreatments (Fig. 4) that expelled the non-cellulosic materials from NC [6]. The peak for NC was well defined and presented a greater lattice peak, symptomatic of the more crystalline nature of NC as compared to the raw OPFL and cellulose. Based on the micrograph, the crystallinity index of NC was estimated to be 45.5%, higher than raw OPFL (25.3%) and cellulose (30.5%). The more crystalline nature of NC was due to the splitting of cellulose glycosidic group into two anhydroglucose units. During the treatment, the amorphic cellulose domains are liberated into the acidic mixture and only the crystalline region of cellulose remains. The results seen here were consistent with the previous findings reported by Dungani et al. [9] and Wulandari et al. [40].

XRD patterns of OPFL, cellulose and NC
3.2.4 Nitrogen Adsorption
Data of adsorption isotherms of cellulose and NC are presented in Fig. 5. As can be seen, cellulose has a surface area of 1.4526 m2/g that was consistent with the larger lateral cellulose dimensions [41]. The value here was somewhat higher than the described surface area for native cellulose (0.6–1.4 m2/g) [42], likely attributable to the difference in sample pretreatment methods and the used gas in the BET technique [42]. Upon acid hydrolysis, the BET surface area of NC was elevated to 5.108 m2/g. Since NC is more crystalline compared to cellulose, the improved surface area of NC was not related to the removal of the amorphous regions of cellulose. This was due to the higher external surface area of NC accompanied by newly formed pores after the acid treatment [43]. The data thus validated that the chemical treatments led to an appreciable increase in the surface area of NC over the cellulose fiber. NCs with higher surface areas have been reported, as the acid hydrolysis process tends to inhibit particles aggregation [44].

BET adsorption isotherms of cellulose and isolated NC
Comparatively, Brinkmann et al. [45] acid-hydrolyzed the kraft pulp using sulfuric acid followed by sodium exchange to produce cellulose nanocrystal (CNC) showing a low surface area (1.290 m2/g) (Table 1). Abu-Danso et al. [44] reported that the BET surface area of CNC extracted from cotton was only 7 m2/g due to an increase in fiber agglomeration. Conversely, Hussin et al. [46] prepared CNC showing a higher surface area (5.085 m2/g) by acid-hydrolyzing the kenaf fiber. Acid hydrolysis of tomato peels by Feng and Hsieh [47] and wood fiber by Wu and co-workers [48] successfully isolated CNC showing surface areas of 21.7 m2/g and 24.12 m2/g, respectively. It is apparent that, despite the same chemical treatment to isolate the CNC from different cellulosic sources, their surface area can differ greatly. This was likely due to variations in soaking time, regularity of stirring and the changing of the acid wash, and possibly environmental temperature during the treatment. Moreover, the low BET surface area of CNC may have been caused by the drying conditions to obtain the nanofibers [50].
3.2.5 Field Emission Scanning Electron Microscopy (FESEM)
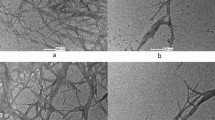
The surface image of NC extracted from raw OPFL after delignification and chemical pretreatments using FESEM is shown in Fig. 6. From the micrographs, the produced NC was uniformly distributed as needlelike structures [28] with diameters ranging between 11 and 28 nm, thus verifying the presence of an entangled network of the nanodimensional NC fibers [51]. The notably coarser surface of NC can be attributed to the dissolution of lignin after the three consecutive chemical treatments. This was similarly described by previous studies by Zhou et al. [52] and Mohaiyiddin et al. [28] which successfully produced NC from microcrystalline cellulose and oil palm fronds, respectively. Based on the findings, the pretreatment protocol carried out in this study was adequate to remove unwanted lignin in the raw OPFL and yielded satisfactory quality of NC.

FESEM micrographs of NC at magnification a 30 000×, b 50 000×, c 60 000× and d 70 000×
Elemental analysis for a specified section of NC was performed using an energy-dispersive X-ray diffraction (EDX) spectrophotometer attached to FESEM, and the results are depicted in Fig. 7. The EDX spectrum (Fig. 7b) revealed peaks around 0.25 keV and 0.50 keV which can be ascribed to the binding energies of carbon and oxygen, respectively. NC was comprised of the elements C and O at 56.7 wt% and 43.3 wt%, respectively, both of which are the main components of the cellulosic fiber without the presence of impurities. The EDX data supported the high purity of the produced NC and correlated well with the FTIR spectra, thus implying the adequacy of the chemical treatments to fully remove hemicellulose and lignin.

a FESEM image of the specified section of NC and b EDX spectrum of NC
3.2.6 Transmission Electron Microscopy (TEM)
The morphological structure of NC was further assessed using TEM, and the results are depicted in Fig. 8. The needlelike structures whose diameters averaged between 10 and 15 nm and showed little agglomeration were similar to the FESEM micrographs. The darker inner side signified that NC particles were embedded onto each other, following the introduction of surface ionic charge after the acid hydrolysis process [3, 53]. The study did, however, observe minor agglomeration that may have occurred when preparing the NC for TEM analysis, particularly when the dispersing medium was removed [11].

TEM images of NC at a 10,000× and b 20,000× magnifications
3.2.7 Atomic Force Microscopy (AFM)
AFM is arguably the most versatile and powerful instrument for studying sample at nanoscale range. In this study, the morphology of NC was further assessed with AFM and the results are depicted in Fig. 9. The dimension of NC produced by the acid hydrolysis process was less than 100 nm with the root-mean-square (RMS) roughness value of 9.146 nm which corresponded quite well with the micrographs of FESEM and TEM. The length of NC that exceeded 100 nm was probably due to aggregation and the stacking of NC onto each other, as previously seen in the FESEM micrographs. In fact, previous studies by Lahiji et al. [54] and Mohaiyiddin et al. [28] have reported a similar occurrence, caused by the formation of intermolecular hydrogen bonds between NC molecules during lyophilization. The apparent darker and brighter regions on NC in the micrographs were indicative of the amorphous and crystalline parts of the nanomaterial, respectively. Since the brighter region appeared more dominant, it can be construed that crystallinity index of NC produced in this study was comparable to that estimated by XRD.

AFM image and 3D image for NC after undergoing a series of chemical treatments
4 Conclusion
NC was successfully extracted from OPFL in the form of whitish powder, and all the non-cellulosic materials, i.e., lignin and hemicellulose, were eliminated after further treatment with an alkali agent. Morphological data obtained from FESEM, TEM and AFM further proved that the synthesis of NC is in the form of their needlelike structures with diameters ranging between 10 and 30 nm. Purity of the NC was supported by the EDX spectrum which revealed no traces of impurities.
Correspondingly, crystallinity of NC was substantially improved over the untreated OPFL fiber and cellulose with a high surface area (5.108 m2/g). In contrary, the cellulose fibers appeared to show marginally enhanced thermal stability compared to NC, as seen from the TGA data due to the insertion of sulfate groups into the cellulose surface during hydrolysis. Marginal changes in the natural structure of cellulose-I polymorph as a result of the chemical pretreatments were further affirmed by the corresponding XRD micrographs.
Hence, we demonstrated that OPFL is a good source of natural fiber to produce NC as a reinforcing agent to yield a more structurally robust alginate composite. Most importantly, this “greener” nanomaterial offers more than a few positive environmental advantages, for instance, raw material utilization, product manufacturing and usefulness until final disposability, as it is completely degradable without liberating hazardous compounds into the environment.
References
Nordin, N.A.; Sulaiman, O.; Hashim, R.; Kassim, M.H.M.: Oil palm frond waste for the production of cellulose nanocrystals. J. Phys. Sci. 28, 115–126 (2017)
Awalludin, M.F.; Sulaiman, O.; Hashim, R.; Wan Nadhari, W.N.A.: An overview of the oil palm industry in Malaysia and its waste utilization through thermochemical conversion, specifically via liquefaction. Renew. Sustain. Energy Rev. 50, 1469–1484 (2015)
Chieng, B.W.; Lee, S.H.; Ibrahim, N.A.; Then, Y.Y.; Loo, Y.Y.: Isolation and characterization of cellulose nanocrystals from oil palm mesocarp fiber. Polymers 9, 355 (2017)
Owolabi, F.A.; Ghazali, A.; Khalil, A.H.P.S.; Hassan, A.; Arjmandi, R.; Fazita, N.M.R.; Haafiz, M.M.K.: Isolation and characterization of microcrystalline cellulose from oil palm fronds using chemomechanical process. Wood Fiber Sci. 48, 1–11 (2015)
Onoja, E.; Chandren, S.; Razak, F.I.A.; Mahat, N.A.; Wahab, R.A.: Oil palm (Elaeis guineensis) biomass in Malaysia: the present and future prospects. Waste Biomass Valorization 10, 2099−2117 (2019)
Onoja, E.; Chandren, S.; Razak, F.I.A.; Wahab, R.A.: Enzymatic synthesis of butyl butyrate by Candida rugosalipase supported on magnetized-nanosilica from oil palm leaves: process optimization, kinetic and thermodynamic study. J Taiwan Inst. Chem. E 91, 105–118 (2018)
Onoja, E.; Chandren, S.; Razak, F.I.A.; Wahab, R.A.: Extraction of nanosilica from oil palm leaves and its application as support for lipase immobilization. J. Biotechnol. 283, 81–96 (2018)
Cherian, B.M.; Leão, A.L.; de Souza, S.F.; Thomas, S.; Pothan, L.A.; Kottaisamy, M.: Isolation of nanocellulose from pineapple leaf fibres by steam explosion. Carbohydr. Polym. 81, 720–725 (2010)
Dungani, R.; Owolabi, A.F.; Saurabh, C.K.; Khalil, A.H.P.S.; Tahir, P.M.; Hazwan, C.I.C.M.; Ajijolakewu, K.A.; Masri, M.M.; Rosamah, E.; Aditiawati, P.: Preparation and fundamental characterization of cellulose nanocrystal from oil palm fronds biomass. J. Polym. Environ. 25, 692–700 (2017)
Elias, N.; Chandren, S.; Razak, F.I.A.; Jamalis, J.; Widodo, N.; Wahab, R.A.: Characterization, optimization and stability studies on Candida rugosa lipase supported on nanocellulose reinforced chitosan prepared from oil palm biomass. Int. J. Biol. Macromol. 114, 306–316 (2018)
Neto, W.P.F.; Silvério, H.A.; Dantas, N.O.; Pasquini, D.: Extraction and characterization of cellulose nanocrystals from agro-industrial residue–Soy hulls. Ind. Crop Prod. 42, 480–488 (2013)
Elias, N.; Chandren, S.; Attan, N.; Mahat, N.A.; Razak, F.I.A.; Jamalis, J.; Wahab, R.A.: Structure and properties of oil palm-based nanocellulose reinforced chitosan nanocomposite for efficient synthesis of butyl butyrate. Carbohydr. Polym. 176, 281–292 (2017)
Abitbol, T.; Rivkin, A.; Cao, Y.; Nevo, Y.; Abraham, E.; Ben-Shalom, T.; Lapidot, S.; Shoseyov, O.: Nanocellulose, a tiny fiber with huge applications. Curr. Opin. Biotechnol. 39, 76–88 (2016)
Khan, A.; Khan, R.A.; Salmieri, S.; Le Tien, C.; Riedl, B.; Bouchard, J.; Chauve, G.; Tan, V.; Kamal, M.R.; Lacroix, M.: Mechanical and barrier properties of nanocrystalline cellulose reinforced chitosan based nanocomposite films. Carbohydr. Polym. 90, 1601–1608 (2012)
Maiti, S.; Jayaramudu, J.; Das, K.; Reddy, S.M.; Sadiku, R.S.S.; Liu, D.: Preparation and characterization of nano-cellulose with new shape from different precursor. Carbohydr. Polym. 98, 562–567 (2013)
Sampath, U.T.M.; Ching, Y.C.; Chuah, C.H.; Singh, R.; Lin, P.C.: Preparation and characterization of nanocellulose reinforced semi-interpenetrating polymer network of chitosan hydrogel. Cellulose 24, 2215–2228 (2017)
Khoo, R.Z.; Ismail, H.; Chow, W.S.: Thermal and morphological properties of poly (lactic acid)/nanocellulose nanocomposites. Procedia Chem. 19, 788–794 (2016)
Li, W.; Wu, Q.; Zhao, X.; Huang, Z.; Cao, J.; Li, J.; Liu, S.: Enhanced thermal and mechanical properties of PVA composites formed with filamentous nanocellulose fibrils. Carbohydr. Polym. 113, 403–410 (2014)
Abraham, E.; Elbi, P.; Deepa, B.; Jyotishkumar, P.; Pothen, L.; Narine, S.; Thomas, S.: X-ray diffraction and biodegradation analysis of green composites of natural rubber/nanocellulose. Polym. Degrad. Stab. 9, 2378–2387 (2012)
Phanthong, P.; Yufei, M.A.; Guan, G.; Abudula, A.: Extraction of nanocellulose from raw apple stem. J. Jpn. Inst. Energy 94, 787–793 (2015)
Navarro-Pardo, F.; Martínez-Barrera, G.; Martínez-Hernández, A.; Castaño, V.; Rivera-Armenta, J.; Medellín-Rodríguez, F.; Velasco-Santos, C.: Effects on the thermo-mechanical and crystallinity properties of nylon 6,6 electrospun fibres reinforced with one dimensional (1D) and two dimensional (2D) carbon. Materials 6, 3494–3513 (2013)
Segal, L.G.J.M.A.; Creely, J.J.; Martin, A.E.; Conrad, C.M.: An empirical method for estimating the degree of crystallinity of native cellulose using the x-ray diffractometer. Text. Res. J. 10, 786–794 (1959)
Rubentheren, V.; Thomas, A.W.; Chee, C.Y.; Tang, C.K.: Processing and analysis of chitosan nanocomposite reinforced with chitin whiskers and tannic acid as a crosslinker. Carbohydr. Polym. 115, 379–387 (2015)
Ching, Y.C.; Ng, T.S.: Effect of preparation conditions on cellulose from oil palm empty fruit bunch fiber. BioResour 9, 6373–6385 (2014)
Malhotra, R.; Prakash, D.; Shukla, S.K.; Kim, T.; Kumar, S.; Rao, N.J.: Comparative study of toxic chlorophenolic compounds generated in various bleaching sequences of wheat straw pulp. Clean Technol Environ. 15, 999–1011 (2013)
Xu, J.; Cheng, J.J.; Sharma-Shivappa, R.R.; Burns, J.C.: Lime pretreatment of switchgrass at mild temperatures for ethanol production. Bioresour. Technol. 101, 2900–2903 (2010)
Lee, H.; Hamid, S.; Zain, S.: Conversion of lignocellulosic biomass to nanocellulose: structure and chemical process. Sci. World J. 2014, 631013 (2014)
Mohaiyiddin, M.S.; Lin, O.H.; Owi, W.T.; Chan, C.H.; Chia, C.H.; Zakaria, S.; Villagracia, A.R.; Akil, H.M.: Characterization of nanocellulose recovery from Elaeis guineensis frond for sustainable development. Clean Technol Environ. 18, 2503–2512 (2016)
Dufresne, A.: Nanocellulose: a new ageless bionanomaterial. Mater. Today 16, 220–227 (2013)
Zhang, J.; Li, B.; Wang, Q.; Wei, X.; Feng, W.; Chen, Y.; Huang, P.; Wang, X.: Application of Fourier transform infrared spectroscopy with chemometrics on postmortem interval estimation based on pericardial fluids. Sci. Rep. 7, 18013 (2017)
Sun, R.C.; Tomkinson, J.; Wang, Y.X.; Xiao, B.: Physico-chemical and structural characterization of hemicelluloses from wheat straw by alkaline peroxide extraction. Polymer 41, 2647–2656 (2000)
Sain, M.; Panthapulakkal, S.: Bioprocess preparation of wheat straw fibers and their characterization. Ind. Crop Prod. 23, 1–8 (2006)
Lani, N.S.; Ngadi, N.; Johari, A.; Jusoh, M.: Isolation, characterization, and application of nanocellulose from oil palm empty fruit bunch fiber as nanocomposites. J. Nanomater. 2014, 702538 (2014)
Alemdar, A.; Sain, M.: Isolation and characterization of nanofibers from agricultural residues—Wheat straw and soy hulls. Bioresour. Technol. 99, 1664–1671 (2008)
Deepa, B.; Abraham, E.; Cordeiro, N.; Mozetic, M.; Mathew, A.P.; Oksman, K.; Faria, M.; Thomas, S.; Pothan, L.A.: Utilization of various lignocellulosic biomass for the production of nanocellulose: a comparative study. Cellulose 22, 1075–1090 (2015)
Troedec, M.; Sedan, D.; Peyratout, C.; Bonnet, J.P.; Smith, A.; Guinebretiere, R.; Gloaguen, V.; Krausz, P.: Influence of various chemical treatments on the composition and structure of hemp fibres. Compos. PartA Appl. Sci. Manuf. 39, 514–522 (2008)
Moriana, R.; Vilaplana, F.; Ek, M.: Cellulose nanocrystals from forest residues as reinforcing agents for composites: a study from macro-to nano-dimensions. Carbohyd. Polym. 139, 139–149 (2016)
Lamaming, J.; Hashim, R.; Leh, C.P.; Sulaiman, O.; Sugimoto, T.; Nasir, M.: Isolation and characterization of cellulose nanocrystals from parenchyma and vascular bundle of oil palm trunk (Elaeis guineensis). Carbohydr. Polym. 134, 534–540 (2015)
Park, S.; Baker, J.O.; Himmel, M.E.; Parilla, P.A.; Johnson, D.K.: Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 3, 10 (2010)
Wulandari, W.T.; Rochliadi, A.; Arcana, I.M.: Nanocellulose prepared by acid hydrolysis of isolated cellulose from sugarcane bagasse. Mater. Sci. Eng. 107, 012045 (2016)
Chunilall, V.; Bush, T.; Larsson, P.T.: Supra-molecular structure and chemical reactivity of cellulose I studied using CP/MAS (sup) 13 C-NMR. Intech Publishing, London (2013)
Vanhatalo, K.M.; Maximova, N.; Perander, A.M.; Johansson, L.S.; Haimi, E.; Dahl, O.: Comparison of conventional and lignin-rich microcrystalline cellulose. BioResour 11, 4037–4054 (2016)
Guo, J.; Catchmark, J.M.: Surface area and porosity of acid hydrolyzed cellulose nanowhiskers and cellulose produced by Gluconacetobacter xylinus. Carbohyd. Polym. 87, 1026–1037 (2012)
Abu-Danso, E.; Srivastava, V.; Sillanpää, M.; Bhatnagar, A.: Pretreatment assisted synthesis and characterization of cellulose nanocrystals and cellulose nanofibers from absorbent cotton. Intl. J. Bio. Macromol. 102, 248–257 (2017)
Brinkmann, A.; Chen, M.; Couillard, M.; Jakubek, Z.J.; Leng, T.; Johnston, L.J.: Correlating cellulose nanocrystal particle size and surface area. Langmuir 32, 6105–6114 (2016)
Hussin, M.H.; Tajudin, N.A.; Azani, N.F.S.M.; Paramasivam, M.; Haafiz, M.K.; Kumar, S.; Yemloul, M.: Physicochemical studies of kenaf nanocrystalline cellulose and poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) as filler for lithium perchlorate based polymer electrolyte. Int. J. Electrochem. Sci. 14, 1620–1633 (2019)
Jiang, F.; Hsieh, Y.L.: Cellulose nanocrystal isolation from tomato peels and assembled nanofibers. Carbohydr. Polym. 122, 60–68 (2015)
Wu, X.; Shi, Z.; Fu, S.; Chen, J.; Berry, R.M.; Tam, K.C.: Strategy for synthesizing porous cellulose nanocrystal supported metal nanocatalysts. ACS Sustain. Chem. Eng. 11, 5929–5935 (2016)
Lu, P.; Hsieh, Y.L.: Preparation and characterization of cellulose nanocrystals from rice straw. Carbohydr. Polym. 87, 564–573 (2012)
Khoshkava, V.; Kamal, M.R.: Effect of drying conditions on cellulose nanocrystal (CNC) agglomerate porosity and dispersibility in polymer nanocomposites. Powder Technol. 261, 288–298 (2014)
Chandra, J.; George, N.; Narayanankutty, S.: K: isolation and characterization of cellulose nanofibrils from arecanut husk fibre. Carbohydr. Polym. 142, 158–166 (2016)
Zhou, Y.M.; Fu, S.Y.; Zheng, L.M.; Zhan, H.Y.: Effect of nanocellulose isolation techniques on the formation of reinforced poly(vinyl alcohol) nanocomposite films. Express Polym. Lett. 6, 794−804 (2012)
Liu, H.; Liu, D.; Yao, F.; Wu, Q.: Fabrication and properties of transparent polymethylmethacrylate/cellulose nanocrystals composites. Bioresour. Technol. 101, 5685–5692 (2010)
Lahiji, R.R.; Xu, X.; Reifenberger, R.; Raman, A.; Rudie, A.; Moon, R.J.: Atomic force microscopy characterization of cellulose nanocrystals. Langmuir 26, 4480–4488 (2010)
Acknowledgements
This work was supported by the Fundamental Research Grant Scheme (FRGS RJ130000.7826.4F871) from the Universiti Teknologi Malaysia, Johor. We would also like to acknowledge the valuable help and suggestions provided by our colleagues.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hussin, F.N.N.M., Attan, N. & Wahab, R.A. Extraction and Characterization of Nanocellulose from Raw Oil Palm Leaves (Elaeis guineensis). Arab J Sci Eng 45, 175–186 (2020). https://doi.org/10.1007/s13369-019-04131-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13369-019-04131-y