
Abstract
Herpes simplex virus type 1 (HSV-1) encephalitis (HSE) is the most common fatal sporadic encephalitis in developed countries. There is evidence from HSE animal models that not only direct virus-mediated damage caused but also the host’s immune response contributes to the high mortality of the disease. Chemokines modulate and orchestrate this immune response. Previous experimental studies in HSE models identified the chemokine receptor CXCR3 and its ligands as molecules with a high impact on the course of HSE in mouse models. In this study, the role of the chemokine receptor CXCR3 was evaluated after intranasal infection with the encephalitogenic HSV-1 strain 17 syn+ using CXCR3-deficient mice (CXCR3−/−) and wild-type controls. We demonstrated a neurotropic viral spread into the CNS of after intranasal infection. Although viral load and histological distribution of infected neurons were independent from CXCR3 signaling early after infection, CXCR3-deficient mice cleared HSV-1 more efficiently 14 days after infection. Furthermore, CXCR3 deficiency led to a decreased weight loss in mice after HSV-1 infection. T cell infiltration and microglial activation was prominently reduced by inhibition of CXCR3 signaling. Quantitative PCR of proinflammatory cytokines and chemokines confirmed the reduced neuroinflammatory response in CXCR3-deficient mice during HSE. Our results demonstrate that the recruitment of peripheral immune cells into the CNS, induction of neuroinflammation, and consecutive weight loss during herpes encephalitis is modulated by CXCR3 signaling. Interruption of the CXCR3 pathway ameliorates the detrimental host immune response and in turn, leads paradoxically to an enhanced viral clearance after intranasal infection. Our data gives further insight into the role of CXCR3 during HSE after intranasal infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Herpes simplex virus type 1 (HSV-1) encephalitis (HSE) is the most common fatal sporadic encephalitis in humans (Whitley and Roizman 2001; Steiner et al. 2007). Treatment with acyclovir decreased its mortality from 70 to 20%. However, only a part of HSE patients fully recover (Sköldenberg et al. 1984; Whitley et al. 1986). It is believed that HSV-1 in humans enters the central nervous system (CNS) via the olfactory pathway or via the trigeminal ganglion (Esiri 1982; Kennedy and Chaudhuri 2002). In mice, development of experimental herpes encephalitis after HSV-1 inoculation is strongly dependent on genetic background and route of infection (Lopez 1975). When administered intranasally (i.n.), HSV-1 enters the CNS and results in an acute necrotizing encephalitis involving the olfactory and limbic systems, including the olfactory bulb, hypothalamus, thalamus, amygdala, hippocampus, and olfactory and entorhinal cortices. Pathological hallmark is a necrotizing encephalitis with primarily HSV-1-infected neurons but also glia (Mori et al. 2005; Shivkumar et al. 2013). The infection induces the host production of proinflammatory cytokines infiltration of macrophages, both CD4+ and CD8+ T lymphocytes, and NK cells into the CNS (Kastrukoff et al. 2010).
The recruitment of immune cells into the lesioned CNS is orchestrated by multiple chemokines during HSV-1 encephalitis (Lundberg and Cantin 2003). Infected mice display a rapid upregulation of several chemokines in the brain, trigeminal ganglion, including CCL2, CCL4, CCL5, CXCL9, and CXCL10. (Carr et al. 1998; Fenton et al. 2002; Cook et al. 2004; Sellner et al. 2005). The receptor for the latter two of these chemokines is CXCR3 (Loetscher et al. 2001). CXCR3 is expressed by activated CD4+ and CD8+ T cells, NK cells, microglia/monocytes, and dendritic cells (Loetscher et al. 1996; García-López et al. 2001). High levels of CXCR3 are found on activated Th1 T cells but not on Th2-polarized cells (Sallusto et al. 1998). The induction of all CXCR3 ligands is mediated by IFN-γ (Luster et al. 1985; Farber 1990). CXCL9 and CXCL10 have been shown to stimulate the chemotaxis of CXCR3+-activated T cells and NK cells in vitro and in several experimental disease models (reviewed in Müller et al. 2010). In addition, there is increasing evidence that CXCR3 is also expressed on resident cells of the CNS, in particular astrocytes, microglia and neurons (Krauthausen et al. 2014; Krauthausen et al. 2015).
The pathomechanistic causes of high mortality and morbidity due to HSV-encephalitis have not been well defined. There is increasing evidence that the host immune response most likely plays a more important role than direct virus induced neuronal damage, e.g., necropsy virus titers in mice do not correlate with mouse resistance or susceptibility genotype (Lundberg et al. 2003; Lundberg et al. 2007), better survival in susceptible mouse strains after depletion of granulocytes and macrophages (Lundberg et al. 2008), long-term MRI abnormalities are reducible in mice by treatment with Acyclovir (ACV) in combination with corticosteroids but not in mice treated with ACV or PBS alone (Meyding-Lamadé et al. 2003). The objective of this study was to further elucidate the role of CXCR3 in experimental HSE. Previous studies in experimental HSE using an ocular infection route suggested that both HSV-1 replication and survival is enhanced in mice deficient for CXCR3 (Lundberg and Cantin 2003; Wickham et al. 2004; Wuest and Carr 2008).
In contrast to previous studies, we further characterized a nasal infection route in HSE and demonstrated an enhanced viral clearance and amelioration of HSE disease course in CXCR3-deficient mice by reduction of detrimental host immune response.
Materials and methods
Animals
CXCR3-deficient (CXCR3KO) mice (originally provided by Drs. Bao Lu and Craig Gerard, Children’s Hospital and Harvard Medical School, Boston, MA) have been described previously (Hancock et al. 2000). The mice were backcrossed at least eight generations onto the C57BL/6 strain. CXCR3-deficient mice displayed no clinical or histologic abnormalities when compared with wild-type (WT) mice.
Experimental procedure
Five-week old mice were anesthetized, and then 10 μL saline containing 106 pfu HSV-1 17 syn+ were injected intranasally. Mock-treated animals served as controls. After infection, mice were examined on a daily basis for clinical signs of HSE and body weight. According to protocol, mice were sacrificed if loss of body weight exceeded more than 30%.
In the first experiment, intranasal route, HSV-infection spread was determined in C57BL/6 wild-type mice. On days 3, 5, 6, 7, 8, and 9, groups of 4–6 mice were perfused transcardially with ice-cold PBS until flow through was completely clear to remove intravascular leukocytes. Brains were removed and cut along the sagittal midline. After overnight fixation, halves of the brains were prepared for paraffin-embedded histology, whereas the other halves where embedded in tissue-tek for fluorescent histology on frozen sections. The whole specimen were cut in 10-μm sagittal serial sections to determine the intracerebral spread of HS-virus infection.
In the second experiment, the influence of CXCR3 signalling was determined on the course of HSE by infecting CXCR3-deficient and wild-type control mice. Again, mock-treated mice of both genotypes served as negative controls. On days 3 (three mice per group), 7, and 14 (6–7 mice per group each), brains were removed after transcardial perfusion with ice-cold PBS. Due to excessive loss of body weight and according to protocol, a HSV-infected wild-type mouse had to be sacrificed on day 9 of the experiment. Brains were cut along the sagittal midline and one half was snap frozen in liquid nitrogen, whereas the other half was fixed overnight in PBS-buffered 4% paraformaldehyde at 4 °C, washed in PBS, and subsequently embedded in paraffin.
Routine histology and immunohistochemistry
Paraffin-embedded sections were stained with H&E and luxol fast blue for routine histological analysis. For immunohistochemistry of paraffin-embedded tissue, sections were rehydrated in graded ethanol series after deparaffination in xylene. Slides were then incubated over night at 4 °C with primary antibodies (rabbit polyclonal anti-HSV-1, 1/100, Dako; rabbit polyclonal anti CD3: 1/200, Dako; Tomato lectin: 1/50, Sigma; monoclonal mouse anti-GFAP: 1:500, Dako). After washing in PBS, a corresponding biotinylated secondary antibody (Axxora, Lörrach, Germany; 1/200) and HRP-coupled streptavidin (Axxora; 1/200) was used. Nova Red (Vector Labs) was applied as the immunoperoxidase substrate according to the manufacturer’s instructions. Sections were counterstained with hematoxylin (Sigma-Aldrich).
Apoptotic neurons on paraffin-sections were detected using TUNEL staining kit (Roche) according to manufacturers protocol with slight modifications.
Fluorescent immunohistochemistry was applied to frozen sections. Primary antibodies were incubated overnight at 4 °C. After washing in PBS, an A594 or A488 fluorescence-conjugated secondary antibody (Invitrogen, Darmstadt, Germany; 1/200, 60 min) was used to visualize the primary antibody. Sections were counterstained with DAPI (Sigma-Aldrich, Munich, Germany). Conventional and immunofluorescence-stained sections were examined under a Nikon eclipse 800 bright-field and fluorescence microscope (Nikon, Düsseldorf, Germany). Bright-field images and monochrome fluorescent images were acquired using a Spot flex camera and SPOT advanced 4.5 software (Diagnostic instruments, Sterling, MI, USA).
Quantitative real-time PCR
RNA of whole brain hemispheres was isolated using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Total RNA (2 μg) was reverse-transcribed into cDNA using SuperScript™ III Reverse Transcriptase (Invitrogen). Real-time quantitative PCR assays were performed using SYBRgreen. The composition of the reaction mixture was as follows: 2 μl of cDNA, 100 nM of each primer, and 1 × SYBR Green PCR Master Mix (Applied Biosystems) in a total volume of 25 μl. Samples were analyzed simultaneously for GAPDH mRNA as the internal control, and quantitative expression values were extrapolated from separate standard curves. Each sample was assayed in duplicate and normalized to GAPDH.
Real-time PCR amplification and detection of HSV-1 DNA was performed on a LightCycler 2.0 instrument (Roche) as described previously in detail (Hafezi et al. 2012).
Statistical analysis
For statistical analysis, GraphPad Prism was used. Weight course was analyzed using a two-way ANOVA followed by a two-tailed Student’s t test with p < 0.05 considered to be statistically significant. Real-time PCR data were analyzed where appropriate by a two-tailed Student’s t test. Due to missing variance in HSV-1 copy numbers on day 14, data were analyzed using a chi-square test with p < 0.05 considered to be statistically significant.
Results
Intranasal HSV-1 inoculation establishes herpes simplex encephalitis with predominant hippocampal viral load and isolated infection of neurons
After intranasal application of HSV-1, all mice developed histological proven herpes simplex encephalitis (HSE). In contrast to other HSE mouse models, clinical severity of HSE is reduced and no spontaneous deaths occurred. Because of weight loss >30%, 3 out of 38 mice had to be sacrificed during disease course (data not shown).
Routine histology revealed extensive anti-HSV-1 staining across the brain of infected mice. Brain regions predominately infected with HSV-1 were the hippocampus (Fig. 1a), basal ganglia (Fig. 1b) and cortex (Fig. 1c). In all areas, anti-HSV-1 staining pattern resembled neuronal morphology. Quantification of HSV-1-infected cells on day 7 using serial sections displayed a clustered distribution of infected neurons in the whole brain. Clusters could be detected, e.g., in the hippocampus, cortex, thalamus, olfactory bulb, brainstem, and cerebellum (Fig. 1d). Immunohistochemistry on frozen sections clearly demonstrated a colocalization of anti-HSV-1 staining and the neuronal marker NeuN (Fig. 1e). In summary, the results of these studies demonstrate an efficient infection of C57BL/6 mice with HSV-1 via intranasal inoculation with low lethality and viral spread into the whole brain.

Intranasal HSV-1 infection results in a widespread HSE with a clustered distribution of infected neurons in the whole brain of wild-type mice. Anti-HSV-1 immunohistochemistry 7 days after intranasal HSV-1 infection revealed a clustered staining pattern especially in neuron-rich brain regions (a–c). Typical predilection sites for HSV-1 infection clusters were the hippocampus (a), thalamus (b), and occipital cortex (c) in wild-type mice. Anti-HSV 1 immunoreactive cells displayed typical morphological criteria for neurons. Semiquantitative analysis of serial sections in six mice demonstrated the hippocampus, cortex, thalamus, hypothalamus, brainstem, and cerebellum to be the most affected brain regions (d; each dot represents the mean number of infected cells in the according brain region). Double immunofluorescent staining for the neuronal marker anti-NeuN (red) and anti-HSV1 (green) clearly identified neurons to be the HSV-1 infected cell population (e)
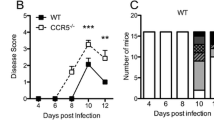
Disruption of CXCR3-signaling ameliorates HSE course after intranasal inoculation
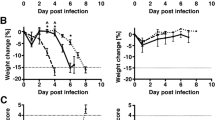
To determine the effect of CXCR3-signaling on the disease course of HSE, CXCR3-deficient mice and wild-type controls were infected using an intranasal route. Body weight was monitored daily and relative changes calculated (Fig. 2). Because of the juvenile age (5 weeks), all experimental groups gained body weight throughout the disease course. Weight gain was impaired in wild-type controls compared to CXCR3-deficient mice (two-way ANOVA p < 0.001). Serial t testing reached statistical significance from day 11 until termination of the experiment on day 14 (p < 0.05). There were no spontaneous deaths in both CXCR3-deficient and wild-type mice. However, a HSV-1-infected wild-type mouse had to be sacrificed on day 9 and removed from the analysis because of more than 30% weight loss. Mock-treated CXCR3-deficient mice and wild-type controls continuously gained about 25% body weight during the experiment without overt differences (data not shown). We also observed less clinical signs in CXCR3-deficient mice like shivering, decrease of movement, and a hunched back. However, these differences were not quantified.

Disruption of CXCR3 signaling protects from weight loss during HSE. Time course of mean relative body weight after intranasal HSV-1 infection in CXCR3-deficient mice (red) and wild-type controls (black) revealed a significantly ameliorated disease course by disruption of CXCR3 signaling (*p < 0.05, dagger removal of one wild-type mouse from experiment after loss of body weight >30%)
Overall, the results of these studies indicated that CXCR3-signaling plays a detrimental role in the clinical course of HSE.
Early neuronal HSV-1 infection is independent from CXCR3-signaling
To determine the influence of CXCR3-signaling on early HSV-1 infection of neurons during HSE, viral load was quantified at 3 and 5 days after disease induction. Histologically viral infection was detected in all brain regions at day 7 with a similar distribution in CXCR3−/− mice and controls (Fig. 3a). Quantification of HSV-1-infected cells on days 3 and 7 after viral inoculation exhibited equal numbers in CXCR3-deficient mice and wild-type controls (Fig. 3b). Furthermore, HSV-1 DNA copy numbers were identical in CXCR3-deficient mice and wild-type controls on days 3 and 7 (Fig. 3c). Taken together, these data demonstrate a CXCR3-independent viral spread into the brain after intranasal HSV-1 infection during HSE.

Early neuronal infection with HSV-1 is independent of CXCR3 signaling. H&E and LFE histology in combination with anti-HSV-1 immunohistochemistry (red) displayed an equal distribution and similar numbers of HSV-1-infected cells in CXCR3-deficient and wild-type mice (a). In wild-type mice, cellular density appeared higher than in CXCR3-deficient mice according to nuclear staining pattern in H&E histology. Semiquantitative analysis of anti-HSV-1 immunohistochemistry in 4–6 mice per group and time point excluded differences in infected cell numbers between CXCR3-deficient and wild-type mice at days 3 and 7 post infection (b). Correspondingly, quantitative PCR of HSV-1 copy numbers could not detect any differences in HSV-1 virus load between CXCR3ko and wild-type mice 3 and 7 days after infection (c, n.d not detectable)
Infiltration of T cells and activation of brain microglia is impaired in CXCR3-deficient mice during HSE
To assess the functional relevance of CXCR3-signaling in neuroinflammation during HSE, we performed immunohistochemistry (Fig. 4). As shown above, numbers of HSV-1-infected neurons did not differ between CXCR3-deficient mice and controls (Fig. 5a, b). As CXCR3 is known to be involved into the recruitment of T cells, we performed anti-CD3 immunohistochemistry 7 days after HSV-1 infection. Whereas numerous T cells were recruited in wild-type mice into HSV-1-infected brain regions (Fig. 5e). T cell numbers remained very low in the brains of CXCR3-deficient mice (Fig. 5f). Furthermore, CXCR3-deficient mice displayed only minor histological signs of microglia activation or macrophage infiltration into these areas compared with wild-type controls shown by lectin staining (Fig. 5c, d). In contrast, anti-GFAP immunohistochemistry revealed pronounced activation of astrocytes by morphological criteria in HSV-1-infected regions of CXCR3−/− mice compared with wild-type controls (Fig. 5g, h).

Reduced T cellular infiltration and microglia/macrophage recruitment but strong astrocytosis by disruption of CXCR3-signaling during HSE. Representative anti-HSV-1 immunohistochemistry 7 days post infection revealed a clustered distribution of HSV-1 infected cells (a, b). Lectin-histochemistry displayed lower numbers of lectin-stained microglia/macrophages in CXCR3ko mice (c, d). Furthermore, anti-CD3 immunohistochemistry in neighboring sections displayed dramatically reduced T-cell numbers in CXCR3-deficient mice (e, f). In contrast, anti-GFAP immunohistochemistry showed a pronounced astrocytosis in HSV-1-infected areas of CXCR3ko mice compared to controls (g, h)

Enhanced viral clearance in CXCR3-deficient mice 14 days after infection. Representative anti-HSV-1 immunohistochemistry of the hippocampus in CXCR3ko and wild-type mice displayed a strong reduction of HSV-1-infected neurons (arrows) (a). Semiquantitative analysis of anti-HSV-1 immunohistochemistry in the whole brain of 5–6 mice per group revealed higher numbers of infected neurons in CXCR3-deficient mice compared with wild-type controls at day 14 post infection without reaching statistical significance (b). Correspondingly, quantitative PCR of HSV-1 copy numbers could not detect any remaining HSV-1 DNA copies in CXCR3-deficient mice compared with high HSV-1 copy numbers in wild type controls (c; n.d. not detectable, p < 0.05)
In summary, these data demonstrate a reduced infiltration of T cells, less activated microglia/macrophages, but pronounced astrocytosis in HSV-1-infected brain regions of CXCR3-deficient mice.
Disrupted CXCR3-signaling facilitates HSV-1 clearance during HSE
To determine the role of CXCR3-signaling on the viral clearance during HSE, HSV-1 load was measured in CXCR3−/− mice and wild-type controls 14 days after infection. Anti-HSV-1 immunohistochemistry revealed an advanced clearance of HSV-1 protein in CXCR3-deficient mice in all areas of the brain, whereas higher numbers of HSV-1-infected cells were still detectable in wild-type controls (Fig. 4a). Visual quantification of HSV-1-infected cells revealed lower numbers of HSV-1-infected cells in CXCR3-deficient mice (Fig. 4b; WT 4.5 ± 1.8 infected cells/visual field vs. CXCR3ko 1.3 ± 0.4 infected cells/visual field). Quantification of HSV-1-DNA copy numbers exhibited a complete clearance of HSV-1 genomic sequences in CXCR3-deficient mice 14 days after infection, whereas HSV-1 DNA in wild-type mice was still detectable (Fig. 4c; WT 1.96 × 106 ± 1.26 × 106 HSV-1 DNA copies/mg vs. CXCR3ko 0 copies/mg, p < 0.05).
Taken together, these results demonstrated a more efficient clearance of Herpes virus from CXCR3-deficient mice compared to wild-type controls 14 days after HSV-1 infection.
Reduced expression of proinflammatory cytokines during HSE in CXCR3-deficient mice
To assess the inflammatory response during HSE in CXCR3-deficient and wild-type mice, we determined the expression of proinflammatory cytokines (Fig. 6). The expression of T cell-produced IFN-γ was upregulated during the whole course of HSE, whereas IFN-γ production in CXCR3-deficient mice remained almost at basal level (day 3 p.i.: WT: 56.3 ± 2.6 vs. CXCR3ko: 3.4 ± 2.4, p < 0.005; day 14 p.i.: WT: 24.5 ± 12 vs. CXCR3ko: 1.3 ± 0.4). Disruption of CXCR3-signaling during HSE resulted in significantly reduced TNF-α production 3 days after infection compared to controls (WT: 10.7 ± 1.2 vs. CXCR3ko: 3.6 ± 1.4, p < 0.05). Fourteen days after infection, TNF-α expression completely normalized in CXCR3-deficient mice, whereas in wild-type animals, TNF-α expression was still detectable (WT: 31.3 ± 18.3 vs. CXCR3ko: 1.1 ± 0.2). Also, the expression of the chemokine CCL2 was induced during the course of HSE with comparable levels at day 3 (WT: 21.0 ± 2.9 vs. CXCR3ko: 11.8 ± 7.9) and higher levels in WT controls at day 14 (WT: 30 ± 12.1 vs. CXCR3ko: 1.4 ± 0.1; p < 0.005). The chemokine CCL5 was highly upregulated during the course of HSE both in CXCR3-deficient and wild-type mice. Fourteen days after disease induction, CCL5 levels peaked in wild-type mice with significant lower levels in CXCR3-deficient mice (WT: 153.6 ± 102.6 vs. CXCR3ko: 8.2 ± 0.7, p < 0.05). The expression of the chemokines CXCL9 and CXCL10 were strongly upregulated 3 days after HSV-1 infection both in wild-type and CXCR3-deficient mice. During the remission phase, 14 days after induction, disruption of CXCR3-signaling resulted in lower expression levels of CXCL9 (WT: 257 ± 123.1 vs. CXCR3ko: 8.2 ± 1.2, p < 0.05) and CXCL10 (WT: 528.5 ± 36.7 vs. CXCR3ko: 4.4 ± 0.6, p < 0.05).

Reduced expression of proinflammatory cytokines and chemokines during HSE in CXCR3-deficient mice. Quantitative PCR of the proinflammatory cytokines and chemokines interferon-γ, TNF-α, CCL2, CCL5, CXCL9, and CXCLl10 3 and 14 days after intranasal HSV-1 infection (white bars: wild type, black bars: CXCR3ko, *p < 0.05)
Taken together, these data demonstrate a strong upregulation of proinflammatory cytokines and chemokines during HSE as described previously. The induction of cytokines was slightly more pronounced in WT controls compared to CXCR3-deficient mice 3 days after infection. These differences reached only a statistical significant level for TNF-α and IFN-γ. Fourteen days after infection, the cytokine levels were strongly diminished in CXCR3-deficient mice compared to wild-type controls.
Discussion
The course of herpes encephalitis is critically modulated by the immune response of the host in response to the viral infection (Steiner et al. 2007; Getts et al. 2008; Steiner and Benninger 2013). The present study examined the course of HSE in CXCR3-deficient mice after infection via the nasal inoculation route. We found an attenuated disease course in CXCR3-deficient mice and surprisingly a more efficient clearance of HSV-1 after 14 days.
In our experimental setting, the nasal infection route featured some advantages over the more commonly used ocular infection. We could establish an infection protocol with a low mortality and prevented severe local symptoms of infection, which would have required the exclusion of the animals due to ethical reasons. In our hands, this was not possible using the ocular infection route.
We characterized the viral spread of HSV-1 after intranasal infection by immunohistochemistry in wild-type mice. As expected, we found clusters of infected neurons in most brain regions, but in particular in the hippocampus, basal ganglia, and cortex, which resembles features of human HSE (Tsalenchuck et al. 2014). To assess herpes virus infection, we focused on quantitative PCR of viral DNA and immunohistochemistry as the number of herpes simplex virus-infected neurons and the number of viral genome copies per neuron correlate with the viral load (Hoshino et al. 2008). These parameters are well established to monitor herpes virus infection burden (Hafezi et al. 2012; Ramakrishna et al. 2015). Concerning the infection of CXCR3-deficient mice, we could not observe differences in the distribution and amount of HSV infection at the early time points examined (day 3 and day 7). This finding suggests that CXCR3 is not critically involved in the initial HSV infection and spread after intranasal infection. However, in contrast to the unimpaired initial infection, we observed a less severe clinical disease course in CXCR3-deficient mice compared to wild-type controls. In particular, an increased weight gain was observed in CXCR3-deficient mice during HSE, which argues for a milder disease course in these mice. As these clinical symptoms are predominantly linked to host’s immune response (Conrady et al. 2010), the attenuated clinical course in CXCR3-deficient mice points toward an important function of CXCR3 in the misdirected immune response against HSV-1. To further characterize and compare the immune response, we detected the infiltration of T cells into the brain parenchyma by immunohistochemistry and found a striking reduction of invading T cells in immunohistochemical analysis. A general role of CXCR3 in T cell infiltration during viral encephalitis is well established. CXCR3 is highly expressed on activated T cells (Farber 1990) and a reduction of invading T cells during viral encephalitis including HSE was reported previously in a plethora of studies (Wickham et al. 2005; Klein et al. 2005; Zhang et al. 2008).
Concerning the effect of CXCR3 on resident cells of the CNS during HSE, we observed diminished activation of microglia in CXCR3-deficient mice by morphological criteria further underlining the potent function of CXCR3 in the host response. The decreased microglial activation could be secondary to the decreased lymphocyte influx or primarily mediated by microglial CXCR3-deficiency. Microglia express a functional relevant CXCR3 receptor and can be modulated by CXCR3 ligands (Biber et al. 2002; Rappert et al. 2002; Krauthausen et al. 2015).
The correlation of a diminished accumulation and activation of immune cells with a less severe disease was described previously (Lundberg et al. 2008) suggesting that this is not a CXCR3-specific observation.
When determining the viral load during HSE, we found a surprisingly complete elimination of HSV in CXCR3-deficient mice compared to wild-type 14 days after infection. The attenuated clinical course in CXCR3-deficient mice during HSE described here was reported previously after ocular infection (Wickham et al. 2005). Though this finding is quite similar to our results, Wickham et al. reported increased viral HSV-1 titers 7 days after infection in CXCR3-deficient mice, which was the latest time-point examined in this study. Using the nasal infection route, we could not confirm this finding at day 7. In contrast at day 14, CXCR3-deficient mice cleared the virus from the brain compared to viral persistence in wild-type mice. Our data supports the view, that a dysregulated immune response during HSE does not only have a negative impact on the clinical course of the disease but may also inhibit an efficient elimination of HSV. Supporting our finding, disruption of CXCR3-signaling has recently been demonstrated to reduce HSV-1 load in brain ependyma, a crucial region for the establishment of encephalitis after ocular infection (Kroll et al. 2014). Kroll et al. showed, similar to our results, an equal progression of HSV-1 into the brainstem 8 days after corneal infection in CXCR3-deficient mice and controls. Interestingly, brain ependyma completely lacked HSV-1 load 8 days after infection in CXCR3-deficient mice. Unfortunately, Kroll et al. could not discriminate between a resistance of CXCR3-deficient brain ependyma and a rapid clearance of the virus within 8 days in this brain region. As we determined the viral load from whole brain lysates, we do not have data regarding the brain ependyma. An IFN-β-mediated mechanism, as proposed by Kroll et al., might be well conceivable.
In summary, CXCR3 critically modulates the course of HSE after nasal infection. CXCR3 is an important molecule for the attraction of immune cells into the infected brain and increases the activation of glial cells. Although, the immune response is diminished in CXCR3-deficient mice, the viral clearance 14 days after infection is strikingly improved compared to wild-type controls. In our hand, CXCR3 signaling contributes to the detrimental host immune response during HSE.
References
Biber K, Dijkstra I, Trebst C et al (2002) Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience 112:487–497
Carr DJJ, Noisakran S, Halford WP et al (1998) Cytokine and chemokine production in HSV-1 latently infected trigeminal ganglion cell cultures: effects of hyperthermic stress. J Neuroimmunol 85:111–121
Conrady CD, Drevets DA, Carr DJJ (2010) Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol 220:1–9. doi:10.1016/j.jneuroim.2009.09.013
Cook WJ, Kramer MF, Walker RM et al (2004) Persistent expression of chemokine and chemokine receptor RNAs at primary and latent sites of herpes simplex virus 1 infection. Virol J 1:5. doi:10.1186/1743-422X-1-5
Esiri MM (1982) Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J Neurol Sci 54:209–226
Farber JM (1990) A macrophage mRNA selectively induced by gamma-interferon encodes a member of the platelet factor 4 family of cytokines. Proc Natl Acad Sci 87:5238–5242. doi:10.1073/pnas.87.14.5238
Fenton RR, Molesworth-Kenyon S, Oakes JE, Lausch RN (2002) Linkage of IL-6 with neutrophil chemoattractant expression in virus-induced ocular inflammation. Invest Ophthalmol Vis Sci 43:737–743
García-López MA, Sánchez-Madrid F, Rodríguez-Frade JM et al (2001) CXCR3 chemokine receptor distribution in normal and inflamed tissues: expression on activated lymphocytes, endothelial cells, and dendritic cells. Lab Investig J Tech Methods Pathol 81:409–418
Getts DR, Balcar VJ, Matsumoto I et al (2008) Viruses and the immune system: their roles in seizure cascade development. J Neurochem 104:1167–1176. doi:10.1111/j.1471-4159.2007.05171.x
Hafezi W, Lorentzen EU, Eing BR et al (2012) Entry of herpes simplex virus type 1 (HSV-1) into the distal axons of trigeminal neurons favors the onset of nonproductive, silent infection. PLoS Pathog 8:e1002679. doi:10.1371/journal.ppat.1002679
Hancock WW, Lu B, Gao W et al (2000) Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med 192:1515–1520
Hoshino A, Nagao T, Nagi-Miura N et al (2008) MPO-ANCA induces IL-17 production by activated neutrophils in vitro via its Fc region- and complement-dependent manner. J Autoimmun 31:79–89. doi:10.1016/j.jaut.2008.03.006
Kastrukoff LF, Lau AS, Takei F et al (2010) Redundancy in the immune system restricts the spread of HSV-1 in the central nervous system (CNS) of C57BL/6 mice. Virology 400:248–258. doi:10.1016/j.virol.2010.02.013
Kennedy PGE, Chaudhuri A (2002) Herpes simplex encephalitis. J Neurol Neurosurg Psychiatry 73:237–238. doi:10.1136/jnnp.73.3.237
Klein RS, Lin E, Zhang B et al (2005) Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol 79:11457–11466. doi:10.1128/JVI.79.17.11457-11466.2005
Krauthausen M, Saxe S, Zimmermann J et al (2014) CXCR3 modulates glial accumulation and activation in cuprizone-induced demyelination of the central nervous system. J Neuroinflammation 11:109. doi:10.1186/1742-2094-11-109
Krauthausen M, Kummer MP, Zimmermann J et al (2015) CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer’s disease model. J Clin Invest 125:365–378. doi:10.1172/JCI66771
Kroll CM, Zheng M, Carr DJJ (2014) Enhanced resistance of CXCR3 deficient mice to ocular HSV-1 infection is due to control of replication in the brain ependyma. J Neuroimmunol 276:219–223. doi:10.1016/j.jneuroim.2014.08.005
Loetscher M, Gerber B, Loetscher P et al (1996) Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med 184:963–969
Loetscher P, Pellegrino A, Gong J-H et al (2001) The ligands of CXC chemokine receptor 3, I-TAC, Mig, and IP10, are natural antagonists for CCR3. J Biol Chem 276:2986–2991. doi:10.1074/jbc.M005652200
Lopez C (1975) Genetics of natural resistance to herpesvirus infections in mice. Nature 258:152–153. doi:10.1038/258152a0
Lundberg P, Cantin E (2003) A potential role for CXCR3 chemokines in the response to ocular HSV infection. Curr Eye Res 26:137–150
Lundberg P, Welander P, Openshaw H et al (2003) A locus on mouse chromosome 6 that determines resistance to herpes simplex virus also influences reactivation, while an unlinked locus augments resistance of female mice. J Virol 77:11661–11673
Lundberg P, Welander PV, Edwards CK 3rd et al (2007) Tumor necrosis factor (TNF) protects resistant C57BL/6 mice against herpes simplex virus-induced encephalitis independently of signaling via TNF receptor 1 or 2. J Virol 81:1451–1460. doi:10.1128/JVI.02243-06
Lundberg P, Ramakrishna C, Brown J et al (2008) The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol 82:7078–7088. doi:10.1128/JVI.00619-08
Luster AD, Unkeless JC, Ravetch JV (1985) γ-Interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature 315:672–676. doi:10.1038/315672a0
Meyding-Lamadé UK, Oberlinner C, Rau PR et al (2003) Experimental herpes simplex virus encephalitis: a combination therapy of acyclovir and glucocorticoids reduces long-term magnetic resonance imaging abnormalities. J Neurovirol 9:118–125. doi:10.1080/13550280390173373
Mori I, Goshima F, Ito H et al (2005) The vomeronasal chemosensory system as a route of neuroinvasion by herpes simplex virus. Virology 334:51–58. doi:10.1016/j.virol.2005.01.023
Müller M, Carter S, Hofer MJ, Campbell IL (2010) Review: the chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity--a tale of conflict and conundrum. Neuropathol Appl Neurobiol 36:368–387. doi:10.1111/j.1365-2990.2010.01089.x
Ramakrishna C, Ferraioli A, Calle A et al (2015) Establishment of HSV1 latency in immunodeficient mice facilitates efficient in vivo reactivation. PLoS Pathog 11:e1004730
Rappert A, Biber K, Nolte C et al (2002) Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol Baltim Md 1950 168:3221–3226
Sallusto F, Lenig D, Mackay CR, Lanzavecchia A (1998) Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med 187:875–883. doi:10.1084/jem.187.6.875
Sellner J, Dvorak F, Zhou Y et al (2005) Acute and long-term alteration of chemokine mRNA expression after anti-viral and anti-inflammatory treatment in herpes simplex virus encephalitis. Neurosci Lett 374:197–202. doi:10.1016/j.neulet.2004.10.054
Shivkumar M, Milho R, May JS et al (2013) Herpes simplex virus 1 targets the murine olfactory neuroepithelium for host entry. J Virol 87:10477–10488. doi:10.1128/JVI.01748-13
Sköldenberg B, Forsgren M, Alestig K et al (1984) Acyclovir versus vidarabine in herpes simplex encephalitis. Randomised multicentre study in consecutive Swedish patients. Lancet 2:707–711
Steiner I, Benninger F (2013) Update on herpes virus infections of the nervous system. Curr Neurol Neurosci Rep 13:414. doi:10.1007/s11910-013-0414-8
Steiner I, Kennedy PGE, Pachner AR (2007) The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6:1015–1028. doi:10.1016/S1474-4422(07)70267-3
Tsalenchuck Y, Tzur T, Steiner I, Panet A (2014) Different modes of herpes simplex virus type 1 spread in brain and skin tissues. J Neurovirol 20:18–27. doi:10.1007/s13365-013-0224-4
Whitley RJ, Roizman B (2001) Herpes simplex virus infections. Lancet 357:1513–1518. doi:10.1016/S0140-6736(00)04638-9
Whitley RJ, Alford CA, Hirsch MS et al (1986) Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med 314:144–149. doi:10.1056/NEJM198601163140303
Wickham S, Lu B, Carr DJJ (2004) The expression of CXCR3 is required for optimal clearance of HSV–1 from the eye and nervous system. Invest Ophthalmol Vis Sci 45:1653–1653
Wickham S, Lu B, Ash J, Carr DJJ (2005) Chemokine receptor deficiency is associated with increased chemokine expression in the peripheral and central nervous systems and increased resistance to herpetic encephalitis. J Neuroimmunol 162:51–59. doi:10.1016/j.jneuroim.2005.01.001
Wuest TR, Carr DJ (2008) Dysregulation of CXCR3 signaling due to CXCL10 deficiency impairs the antiviral response to herpes simplex virus 1 infection. J Immunol 181:7985–7993
Zhang B, Chan YK, Lu B et al (2008) CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J Immunol Baltim Md 1950 180:2641–2649
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
J. Zimmermann and W. Hafezi contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zimmermann, J., Hafezi, W., Dockhorn, A. et al. Enhanced viral clearance and reduced leukocyte infiltration in experimental herpes encephalitis after intranasal infection of CXCR3-deficient mice. J. Neurovirol. 23, 394–403 (2017). https://doi.org/10.1007/s13365-016-0508-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-016-0508-6