
Abstract
The neuropathogenesis of HIV-associated neurocognitive disorders (HAND) remains puzzling. We interrogated several levels of data (host genetic, histopathology, brain viral load, and neurocognitive) to identify histopathological changes most relevant to HAND. The design of the study is a clinicopathological study employing genetic association analyses. Data and brain tissue from 80 HIV-infected adults were used. Markers in monocyte chemoattractant protein-1 (MCP-1), interleukin 1-alpha (IL1-α), macrophage inflammatory protein 1-alpha (MIP1-α), DRD3, DRD2, and apolipoprotein E (ApoE) were genotyped. Microtubule associated protein 2 (MAP2), synaptophysin (SYP), human leukocyte antigen-DR (HLA-DR), glial fibrillary acidic protein (GFAP), amyloid beta (A-Beta), and ionized calcium-binding adaptor molecule-1 (Iba-1) immunoreactivity were quantified in the frontal cortex, putamen, and hippocampus. A composite score for each marker (mean of the three brain regions) was used. Neurocognitive functioning and other clinical variables were determined within 1 year of death. Brain HIV RNA viral load was available for a subset of cases. MAP2 and SYP proved most relevant to neurocognitive functioning. Immunoreactivity of these markers, as well as A-Beta and Iba-1, was correlated with brain HIV RNA viral load. Several genetic markers in combination with other factors predicted histopathology: HIV blood viral load, MIP1-α genotype, and DRD3 genotype predicted Iba-1 immunoreactivity; the duration of infection and IL1-α genotype predicted GFAP immunoreactivity; ApoE genotype and age at death predicted A-Beta immunoreactivity. These data indicate that HIV replication in the brain is the primary driving force leading to neuroinflammation and dysfunctional protein clearance, as reflected by A-Beta and Iba-1. Downstream to these changes are synaptodendritic degeneration, which is the immediate histopathological substrate of the neurocognitive impairment characteristic of HAND. These intermediate histopathological phenotypes are influenced by host genetic polymorphisms in genes encoding cytokines/chemokines, neuronal protein clearance pathways, and dopaminergic factors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pharmaceutical advances built upon immunologic and genetic research have greatly improved the lives of HIV-infected (HIV+) individuals. Despite this, the prevalence of HIV-associated neurocognitive disorders (HAND) remains largely unchanged, although it is generally less severe (Heaton et al. 2011). Prior to the development of combination antiretroviral therapy (cART), the most severe form of HAND, HIV-associated dementia (HAD), was more common. Then, as now, HIV encephalitis (HIVE) was considered to be a major neuropathological basis of HAD (McArthur et al. 1993; Moore et al. 2006; Glass et al. 1995; Bell et al. 1998; Persidsky and Gendelman 2003; Everall et al. 2005; Letendre et al. 2011; Boven et al. 2000; Conant et al. 1998; Eugenin et al. 2006; Kraft-Terry et al. 2009). However, in the current era of widespread cART use the vast majority of HAND cases present with milder symptoms (McArthur et al. 2005, Heaton et al. 2011) and do not have neuropathological findings consistent with HIVE (Everall et al. 2009). Accumulating evidence suggests that for the vast majority of cART-era HAND cases, which are mild-to-moderate in severity, the neuropathogenesis of HAND is due largely to neurodegeneration driven by chronic neuroinflammation (Glass et al. 1995; Kraft-Terry et al. 2009; Persidsky and Gendelman 2003; Everall et al. 2009), but the specific neuropathological markers involved are unclear. Putative markers include synaptophysin and microtubule-associated protein-2 (Moore et al. 2006). abnormal protein aggregation such as β-amyloid (Achim et al. 2009; Rempel and Pulliam 2005; Green et al. 2005; Esiri et al. 1998; Soontornniyomkij et al. 2012). and a variety of histopathological markers reflecting macrophage proliferation, microglial activation, astroglial activation, and dysregulated cytokine expression and production (Bell et al. 1998; Glass et al. 1995; Persidsky and Gendelman 2003; Everall et al. 2005; Letendre et al. 2011; Boven et al. 2000; Conant et al. 1998; Eugenin et al. 2006; Kraft-Terry et al. 2009). Determining the relative importance of these various neuropathological indicators would advance our understanding of HAND pathogenesis.
Candidate gene studies have identified variants within immune-related genes that modify risk for HAND (as reviewed in (Levine et al. 2014a; Kallianpur and Levine 2014)). As one would suspect based on neuropathological studies, these largely involve immune factors involved in neuroinflammatory (Letendre et al. 2011). including cytokines, chemokines, and their receptors. It is well-established that chemokines can serve to block HIV-1 co-receptors thereby modifying HIV replication (Lane et al. 2003) and disease progression (Gonzalez et al. 2001, 2005). as well as influence macrophage activation and chemotaxis of monocytes and other cells across the blood-brain barrier (Kaul and Lipton 2005; Weiss et al. 1999). thus leading to increased inflammation and viral seeding of the CNS. Chemokines also affect neuronal signaling with subsequent disturbance of glial and neuronal functions (Zheng et al. 2001). Immunologic factors germane to HAND include monocyte chemoattractant protein-1 (MCP-1) (Conant et al. 1998; Kelder et al. 1998; Letendre et al. 2011; Lehmann et al. 2006). macrophage inflammatory protein 1-alpha (MIP1-α) (Schmidtmayerova et al. 1996). apolipoprotein E (ApoE) (Vitek et al. 2009). and interleukin 1-alpha (IL-1α) (Nolting et al. 2009). In addition to immune factors, candidate gene studies have also implicated dopaminergic dysregulation in HAND neuropathogenesis (Kumar et al. 2009, 2011; Levine et al. 2012, 2014a, b). However, candidate gene studies thus far have almost exclusively utilized behavioral phenotypes (e.g., HAND diagnosis or neurocognitive functioning), perhaps explaining the variable results in validation studies.
If host genetic variants do modify risk for HAND, it logically follows that they must also modify the cellular and pathophysiological pathways that underlie HAND. While this approach has recently been applied in the study of Alzheimer’s disease (Mortimer et al. 2009; Bennett et al. 2005; Shulman et al. 2010; Bennett et al. 2009). it remains unexplored in the context of HAND. Bridging the informational gap between genotype and behavioral phenotype in the context of HAND may provide important insights about pathogenesis. In this study, we have quantified several histopathological markers deemed relevant to HAND and have genotyped genes variably shown to be associated with risk of HAND or neurocognitive impairment in the context of HIV. In addition, we have obtained brain HIV RNA viral load from the brains of a subset of individuals. Using these data, this multilevel analysis determined the relationship between genetic susceptibility loci, histopathological markers, and brain HIV viral load in leading to neurocognitive impairment.
Methods
Tissue source
This study was conducted in accordance with the University of California, Los Angeles Medical Institutional Review Board. Data and biological samples came from 80 individuals enrolled in either the National Neurological AIDS Bank (NNAB) or California NeuroAIDS Tissue Network (CNTN) who died between August 1999 and January of 2012. For inclusion, all cases were required to be HIV+, over age 18, and have been diagnosed as either neurocognitively normal or with HAND within 1 year prior to death, per established research criteria (Antinori et al. 2007; The Dana consortium on therapy for HIV dementia and related cognitive disorders 1996). All cases died well into the cART era (post-1996). Exclusion criteria were (1) pre- or postmortem evidence of non-HIV-related neurological diseases (e.g., stroke, neoplasm, multiple sclerosis, traumatic brain injury, and neurodegenerative illness), (2) history or evidence of toxoplasmosis or progressive multifocal leukoencephalopathy, and (3) diagnosis of substance dependence within 1 year before death. Comorbid medical conditions, largely self-reported by participants and not recorded in a standardized way when these cases were alive, were available for up to 75 % of the sample, depending on the condition. Sample characteristics are shown in Tables 1 and 2.
Clinical variables
HAND severity
The diagnosis of HAND and determination of severity were accomplished via multidisciplinary consensus based on either the previous 1996 American Academy of Neurology (AAN) criteria if their last premortem evaluation occurred prior to the publication of the newer HAND research criteria “Frascati” in 2007 or according to the Frascati criteria if after 2007. The NNAB and CNTN diagnostic criteria prior to 2007 included a “subsyndromic” impairment classification that is essentially identical to asymptomatic neurocognitive impairment according to the Frascati criteria. Because the criteria for mild cognitive/motor disorder and HAD (per AAN criteria) remained essentially identical to mild neurocognitive disorder and HAD (per Frascati: Antinori et al. 2007 criteria), respectively, we were able to group participants from both diagnostic eras. For this study, levels of severity were neurocognitively normal, asymptomatic neurocognitive impairment, mild neurocognitive disorder, and HAD.
Neurocognitive functioning
Neuropsychological clinical ratings were determined for each case based on neurocognitive test scores obtained within 1 year of death. The clinical ratings approach uses demographically corrected T-scores from a comprehensive neuropsychological battery that are categorized by domain of cognitive functioning, as previously described (Woods et al. 2004). Clinical ratings for each domain were assigned on a scale that ranged from 1 (above average) to 9 (severely impaired), with scores of 5 or more indicative of impairment. These were summarized as a global clinical rating (GCR). Among individuals living with HIV, GCR is associated with daily functioning abilities (Heaton et al. 2011). HIV disease variables (Heaton et al. 2011). and synaptodendritic changes (Moore et al. 2006).
HIV disease measures
Peripheral blood was collected from living participants by venipuncture into EDTA and heparinized tubes prior to death and was assayed for HIV RNA viral load using the Roche Amplicor Assay and by flow cytometry for CD4+ T-lymphocyte subsets. HIV plasma viral load was measured at the last premortem visit within 1 year of death. Duration of HIV infection (based on self-report) and nadir CD4+ cell count (often self-reported) were also recorded. For a subset of cases, HIV RNA levels measured in mid-frontal cortex were available, described below. Plasma measures of viral load and CD4+ cell counts were not available at the time of death because venipuncture cannot be collected after the heart has ceased beating due to blood coagulation.
Antiretroviral CNS penetration or effectiveness
We employed the CNS penetration or effectiveness (CPE), a score that is based on the pharmacologic characteristics of antiretroviral medications (Letendre 2011). The CPE of individual antiretroviral drugs is ranked from 1 (poorest) to 4 (best) based on the 2010 ranking system (Letendre et al. 2010). The CPE score for each case was derived by adding ranks of all antiretroviral drugs within their regimen, which was reported at the time of neurocognitive testing. Higher scores indicate a regimen with increased penetration of the blood brain barrier.
Brain HIV RNA viral load
For a subset of 39 cases, HIV RNA quantified from the dorsolateral frontal cortex as part of a separate study and made available for analysis here (Gelman et al. 2013). Details are provided in the Supplementary materials.
Tissue processing, DNA extraction, and genotyping
Frozen occipital cortex samples were shipped to the University of California Los Angeles—Biological Samples Processing Core from the NNAB and CNTN for DNA extraction. The Autopure LS nucleic acid purification instrument was used for extracting DNA. DNA extraction and genotyping methods are provided in the Supplementary materials. The following genetic markers were examined in this study: MCP-1 (rs1024611), MIP1-α (rs1719134), DRD3 (rs6280), DRD2 (rs6277), ApoE (rs429358), and IL1-α (rs17561).
Histopathological characterization
Histopathological characterization was accomplished using previously described methods (Masliah et al. 1997; Moore et al. 2006) based on immunohistochemistry and conducted on fixed/frozen vibratome sections or formalin-fixed paraffin-embedded sections. Right dorsolateral mid-frontal cortex, hippocampus, and putamen were obtained from deceased HIV+ patients as soon as possible after death and either frozen or fixed in formalin. For the statistical analyses, we generally used a composite score, which was the mean value from the three regions, for each of the following markers: synaptophysin (SYP), microtubule associated protein 2 (MAP2), human leukocyte antigen-DR (HLA-DR), ionized calcium-binding adaptor molecule-1 (Iba-1), glial fibrillary acidic protein (GFAP), and amyloid beta (A-Beta). MAP2 and SYP were available only for the 35 from the CNTN. Additional details of histopathological analysis are in the Supplementary materials.
Statistical analysis
As a preliminary step, we examined the role of comorbid medical conditions (present or not present) on neurocognitive functioning and histopathology using ANOVA. Any comorbid medical conditions found to influence these outcomes were included in later analyses. Comorbid conditions in which fewer than five individuals reported the illness were not considered.
We then sought to examine the relevance of histopathological markers to HAND. This was accomplished by correlating the various markers with GCR and by comparing the immunoreactivity for each marker between those diagnosed with HAND and those considered neurocognitively normal at their final evaluation within 1 year of death. Because we had quantified these markers across several brain regions, we were able to examine both global neurocognitive functioning and domain-specific associations (e.g., memory and histopathology in the hippocampus). Nonparametric statistical tests were used (Spearman’s rho and Kruskal-Wallis).
Having quantified several histopathological markers across several brain regions, we then examined the relationship among these markers. A composite score (mean value across the frontal cortex, putamen, and hippocampus) was calculated for each marker, and that value was correlated across brain regions via Spearman’s rho.
For 39 cases, we examined the relationship between HIV RNA levels, histopathological markers, and host genotype using Pearson and Spearman’s rho correlation tests.
Finally, we then sought to determine the effect of genotype on extent of histopathology. This was accomplished with Kruskal-Wallis for MAP2 and SYP (due to the small number of available samples) and linear regression for GFAP, Iba1, HLA-DR, and A-Beta. A separate regression was run for a composite measure of each marker (averaged across the three brain regions), with the marker as the dependent variable and several predictor variables entered as follows: Block (1) age at death, race, gender, education, and relevant comorbid medical conditions; Block (2) duration of HIV infection, nadir CD4+ cell count, log10 blood HIV RNA viral load; Block (3) genotypes (as described above, entered in a forward stepwise manner). Note that CPE scores were not available for several cases. Therefore, the regression analyses were completed without this variable. When significant results were found, we repeated the regression with CPE. For MAP2 and SYP, due to the small number of cases with those markers characterized, genotype group differences in median values were determined for each gene using the Kruskal-Wallis test. Multiple comparisons for most analyses were corrected with the false discovery rate (FDR) (Benjamini et al. 2001) method.
Results
Relationship of comorbid medical conditions with histopathological markers and neurocognitive functioning
No group differences were observed after correcting for multiple testing. Results are available in the Supplementary materials.
Relationship between histopathological markers and neurocognitive functioning or HAND
Both MAP2 and SYP were significantly correlated with global neurocognitive functioning and HAND severity. Secondary analyses showed this to be true across virtually all brain regions and cognitive domains (Table 3), although this latter analysis was not corrected for multiple comparisons. No clear pattern was observed relating the regional immunoreactivity of MAP2 or SYP to particular neurocognitive domains.
Marginal correlations were observed between A-Beta in putamen and attention/working memory (r = 0.25, p = .037), GFAP in putamen with motor (r = 0.24, p = .034), and HLA-DR in frontal cortex with learning (r = 0.27, p = .017). These correlations were not significant after correction using the FDR (Benjamini et al. 2001).
Relationship between histopathological markers and HIV RNA
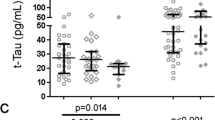
We next examined the relationship between histopathological markers in frontal cortex with HIV RNA. Again, the most robust correlations were observed between HIV RNA and both MAP2 (r = −0.60, p = .006) and SYP (r = −0.58, p = .01). In addition, both Iba1 and A-Beta were significantly correlated with HIV RNA in the frontal cortex (Table 4).
Relationship among histopathological markers
Correlations among the various histopathological markers were examined separately within the frontal cortex, putamen, and hippocampus. Results are shown in the Supplementary materials. MAP2 and SYP were moderately to strongly correlated across all three regions (frontal: r = 0.61, p < .0001; putamen: r = 0.85, p < .0001; hippocampus: r = 0.68, p < .0001). Interestingly, we also observed moderate correlations between GFAP and Iba1 across all three regions (frontal: r = 0.40, p = .0002; putamen: r = 0.46, p < .0001; hippocampus: r = 0.55, p < .0001). SYP and A-Beta were negatively correlated in frontal cortex (r = −0.55, p = .001) and putamen (r = −0.39, p = .029). Note that this second correlation did not survive FDR correction.
Relationship between genotypes and histopathological markers
As described above, the number of samples with MAP2 and SYP was too small to conduct regression analyses. Therefore, potential differences in MAP2 and SYP immunoreactivity between genotype groups were examined with the Kruskal-Wallis test. No significant differences in MAP2 or SYP immunoreactivity were observed between genotype groups for any of the markers examined.
Linear regression was used to examine the remaining histopathological markers. Only the models without CPE are shown in Table 5. For HLA-DR, the regression model was not significant (F = .564, p = .757, adjusted R 2 = −.045). The Iba-1 model was significant (F = 2.813, p = .01, adjusted R 2 = .192), with significant predictors being log10 blood HIV RNA viral load (p = .03), MIP1α genotype (p = .005), and DRD3 genotype (p = .036). The regression for GFAP was also significant (F = 4.787, p = .0003, adjusted R 2 = .303), with duration of infection (p = .007) and IL-1α genotype (p = .0002) as significant predictors. Finally, the model for A-Beta was significant (F = 2.474, p = .028, adjusted R 2 = .145), with ApoE genotype as a significant predictor (p = .048) and trend for age at death (p = .052).
All significant models were rerun with CPE, which reduced the sample size to 48. While underpowered, the results were similar to the models that did not include CPE, with the exception that age at death, which became a significant predictor in the A-Beta model.
Discussion
HAND is a multifactorial syndrome because, while it is attributed to HIV infection, it is also influenced by a wide range of other factors (Martin-Thormeyer and Paul 2009; Cohen and Gongvatana 2010; Gonzalez and Cherner 2008; Brown et al. 2014). As with susceptibility for HIV infection and rate of disease progression, there appears to be several genes that modify the risk for HAND and its course. It has been difficult to validate these genetic association studies, likely due largely to the use of different diagnostic schema or endophenotypes across studies (Woods et al. 2004; Levine et al. 2014a). Furthermore, because the pathogenesis of HAND is likely multifactorial, and HAND itself varies in presentation and course (Lojek and Bornstein 2005; Dawes et al. 2008; Fazeli et al. 2014). it is possible that several genes serving various underlying processes are involved. Therefore, to understand HAND pathogenesis, a strategy that encompasses several levels of inquiry is required. In this study, we have interrogated quantitative measures of six histopathological markers, several relevant host genotypes, neurocognitive functioning, and brain HIV RNA viral load in an effort to understand the interrelationship among these layers of information and create a causative model of HAND pathogenesis.
Our findings indicate that the histopathological markers most strongly related to HAND are MAP2 and SYP. These markers reflect synaptodendritic integrity and demonstrate robust correlations with global premortem neurocognitive functioning, as demonstrated in previous studies (Moore et al. 2006; Masliah et al. 1997). Other histopathological markers, including those reflecting microglial and macrophage proliferation, astrogliosis, and impaired protein clearance (A-Beta), were not correlated with neurocognitive functioning or HAND severity in this sample. It is also notable that of the 20 cases diagnosed as HAD within 1 year prior to death, only one had HIVE.
Based on a subset of cases, it appears that brain HIV RNA viral load is the catalyst of HAND-related histopathological changes. This was most clear with MAP2 and SYP; as HIV RNA viral load increased, synaptodendritic integrity within the same region decreased. While this is based on correlation analysis, it is most logical to conclude a unidirectional effect (viral load → histopathology), save for the possibility of another unmeasured factor. Synaptodendritic degeneration associated with HIV RNA viral abundance has long been suspected as a basis of HAND, even prior to cART use (Masliah et al. 1992). It is also notable that as HIV RNA increased, there was a concomitant increase in A-Beta and Iba1, suggesting dysfunctional protein clearance and neuroinflammation. Furthermore, the associations among the histopathological markers indicated a strong correlation between MAP2 and SYP across all regions and an inverse relationship between SYP and A-Beta particularly in frontal cortex. This raises the question of whether HIV replication in the brain was driving all of the aforementioned histopathological changes or if it initiated a causal chain of events involving multiple histopathological changes.
Our final analyses considered how demographic factors, HIV disease variables, and host genotype predicted histopathology in the brain. Due to the lower number of cases with MAP2 and SYP, it was not possible to examine these markers via linear regression, but genotype group comparisons did not reveal differences in MAP2 or SYP immunoreactivity. However, three of the remaining four histopathological markers were influenced by genotype, and the findings are biologically coherent. First, immunoreactivity of Iba1, a marker of microglial and macrophage proliferation, increased as a function of premortem blood HIV RNA viral load, DRD3 genotype, and MIP1-α genotype. In this model, elevated blood viral load may reflect more active replication in the CNS, leading to increased neuroinflammation (brain viral load was not included in the regression analyses due to limited availability of those data). The contribution of genotype is less straightforward. The DRD3 C (rs6280) allele has been previously associated with poorer neurocognitive functioning (Bombin et al. 2008; Gupta et al. 2011). The A allele of MIP1-α (rs1719134) has been linked to faster HIV disease progression (Gonzalez et al. 2001) and is in high linkage disequilibrium with a single nucleotide (rs11370771) (Modi et al. 2006) previously associated with HAD (Levine et al. 2009). It is unclear what the functional consequences of these polymorphisms are, but MIP-1α is a potent macrophage chemoattractant and natural ligand for the HIV co-receptor CCR5. Our findings suggest that this polymorphism is associated with immunologic response to increasing viral replication, leading to increased migration of monocytes into the CNS and subsequent neuroinflammation. Secondly, we found increasing GFAP immunoreactivity as a function of HIV infection duration and possession of one or two IL-1α T alleles. IL-1α is a pro-inflammatory factor produced primarily by macrophages, and this polymorphism has been linked to the onset of multiple sclerosis and other diseases (Charbonneau et al. 2014; Mirowska-Guzel et al. 2011) and was previously associated with poor control of plasma HIV viremia (Price et al. 2004). however, previous association studies have not linked this polymorphism to HAND (Pemberton et al. 2008; Levine et al. 2009, 2014a). GFAP was not correlated with MAP2 or SYP (Supplementary materials). Together, these findings suggest that while astrogliosis increases with duration of infection and inflammatory response, it may not be the relevant neuropathological process underlying HAND. Finally, A-Beta levels increased as a function of age at death and possession of one or two ApoE-ɛ4 alleles. Additionally, the strong correlation between A-Beta and MAP2/SYP (Supplementary materials) suggests that this histopathological change may be relevant to HAND. As a whole, these findings indicate that active HIV replication in the brain is the primary driving force leading to neuroinflammation (Iba1) and dysfunctional protein clearance (A-Beta), and that downstream to these changes is synaptodendritic degeneration which is the immediate histopathological substrate of HAND, although several factors modify this cascade (Lu et al. 2011; Hinkin et al. 2008; Lamers et al. 2011).
The conclusions should be considered with the following caveats. Firstly, while our sample size was generally adequate for most analyses, we were unable to include some relevant variables (e.g., CPE). The small sample also precluded us from conducting stratified analyses based on ethnicity, as phenomena such as population stratification can confound genetic association studies; however, we included ethnicity in the regression models. Secondly, several variables were collected up to 12 months prior to death, leaving open the possibility that they were no longer applicable. This is unavoidable in clinicopathological studies such as this. Thirdly, our sample was derived from cohorts that historically recruited individuals with advanced illness (i.e., AIDS). This is reflected in the relatively high rate of HAND, in particularly HAD, as well as the young average age at death. Thus, the generalizability of our findings in the current HAND era may be somewhat dampened. Finally, cause of death, which may have an effect on histopathology, was largely indeterminable in the majority of cases, and this information is not available through the NNTC. However, we did exclude cases with clear neuropathological indicators of neurological-related causes of death (e.g., stroke).
Understanding the neuropathogenesis of complex syndromes such as HAND requires a multilevel approach anchored in genotype and utilizing reliable phenotypes across layers. In the present study, we have presented a preliminary, data-supported model that indicates the influence of HIV replication on various histopathological and their subsequent influence on neurocognitive functioning, and how this relationship is modified by host genotype. As we further develop this ambitious model to bridge genetic, neuropathology, and clinical outcomes, a comprehensive understanding of HAND pathogenesis will emerge.
References
Achim CL, Adame A, Dumaop W, Everall IP, Masliah E (2009) Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J Neuroimmune Pharmacol 4:190–199
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, Mcarthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE (2007) Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69:1789–1799
Bell JE, Brettle RP, Chiswick A, Simmonds P (1998) HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain 121(Pt 11):2043–2052
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I (2001) Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125:279–284
Bennett DA, Schneider JA, Wilson RS, Bienias JL, Berry-Kravis E, Arnold SE (2005) Amyloid mediates the association of apolipoprotein E e4 allele to cognitive function in older people. J Neurol Neurosurg Psychiatry 76:1194–1199
Bennett DA, DE Jager PL, Leurgans SE, Schneider JA (2009) Neuropathologic intermediate phenotypes enhance association to Alzheimer susceptibility alleles. Neurology 72:1495–1503
Bombin I, Arango C, Mayoral M, Castro-Fornieles J, Gonzalez-Pinto A, Gonzalez-Gomez C, Moreno D, PARELLADA M, Baeza I, Graell M, Otero S, Saiz PA, Patino-Garcia A (2008) DRD3, but not COMT or DRD2, genotype affects executive functions in healthy and first-episode psychosis adolescents. Am J Med Genet B Neuropsychiatr Genet 147B:873–879
Boven LA, Middel J, BREIJ EC, Schotte D, Verhoef J, SODERLAND C, Nottet HS (2000) Interactions between HIV-infected monocyte-derived macrophages and human brain microvascular endothelial cells result in increased expression of CC chemokines. J Neurovirol 6:382–389
Brown LA, Jin J, Ferrell D, Sadic E, Obregon D, Smith AJ, Tan J, Giunta B (2014) Efavirenz promotes beta-secretase expression and increased Abeta1-40,42 via oxidative stress and reduced microglial phagocytosis: implications for HIV associated neurocognitive disorders (HAND). PLoS One 9:e95500
Charbonneau B, Block MS, Bamlet WR, Vierkant RA, Kalli KR, Fogarty Z, Rider DN, Sellers TA, Tworoger SS, Poole E, Risch HA, Salvesen HB, Kiemeney LA, Baglietto L, Giles GG, Severi G, Trabert B, Wentzensen N, Chenevix-Trench G, For A. A. C. S. G, Whittemore AS, Sieh W, Chang-Claude J, Bandera EV, Orlow I, Terry K, Goodman MT, Thompson PJ, Cook LS, Rossing MA, Ness RB, Narod SA, Kupryjanczyk J, Lu K, Butzow R, Dork T, Pejovic T, Campbell I, Le ND, Bunker CH, Bogdanova N, Runnebaum IB, Eccles D, Paul J, Wu AH, Gayther SA, Hogdall E, Heitz F, Kaye SB, Karlan BY, Anton-Culver H, Gronwald J, Hogdall CK, Lambrechts D, Fasching PA, Menon U, Schildkraut J, Pearce CL, Levine DA, Kjaer SK, Cramer D, Flanagan JM, Phelan CM, Brown R, MASSUGER LF, Song H, Doherty JA, Krakstad C, Liang D, Odunsi K, Berchuck A, Jensen A, Lubinski J, Nevanlinna H, Bean YT, Lurie G, Ziogas A, Walsh C, Despierre E, Brinton L, Hein A, Rudolph A, Dansonka-Mieszkowska A, Olson SH, Harter P, Tyrer J, Vitonis AF, Brooks-Wilson A, Aben KK, Pike MC, Ramus SJ, Wik E, Cybulski C, Lin J, Sucheston L, Edwards R, Mcguire V, Lester J, Du Bois A, Lundvall L et al (2014) Risk of ovarian cancer and the NF-kappaB pathway: genetic association with IL1A and TNFSF10. Cancer Res 74:852–861
(1996) Clinical confirmation of the American Academy of Neurology algorithm for HIV-1-associated cognitive/motor disorder. The Dana consortium on therapy for HIV dementia and related cognitive disorders. Neurology 47, 1247–53. Available at: http://www.ncbi.nlm.nih.gov/pubmed/?term=The+Dana+consortium+on+therapy+for+HIV+dementia+and+related+cognitive+disorders+and+1996
Cohen RA, Gongvatana A (2010) The persistence of HIV-associated neurocognitive dysfunction and the effects of comorbidities. Neurology 75:2052–2053
Conant K, Garzino-Demo A, Nath A, Mcarthur JC, Halliday W, Power C, Gallo RC, Major EO (1998) Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A 95:3117–3121
Dawes S, Suarez P, Casey CY, Cherner M, Marcotte TD, Letendre S, Grant I, Heaton RK (2008) Variable patterns of neuropsychological performance in HIV-1 infection. J Clin Exp Neuropsychol 30:613–626
Esiri MM, Biddolph SC, Morris CS (1998) Prevalence of Alzheimer plaques in AIDS. J Neurol Neurosurg Psychiatry 65:29–33
Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW (2006) CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci 26:1098–1106
Everall IP, Hansen LA, Masliah E (2005) The shifting patterns of HIV encephalitis neuropathology. Neurotox Res 8:51–61
Everall I, Vaida F, Khanlou N, Lazzaretto D, Achim C, Letendre S, Moore D, Ellis R, Cherner M, Gelman B, Morgello S, Singer E, Grant I, Masliah E, National Neuro, A. T. C (2009) Cliniconeuropathologic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol 15:360–370
Fazeli PL, Crowe M, Ross LA, Wadley V, Ball K, Vance DE (2014) Cognitive functioning in adults aging with HIV: a cross-sectional analysis of cognitive subtypes and influential factors. J Clin Res HIV AIDS Prev 1:155–169
Gelman BB, Lisinicchia JG, Morgello S, Masliah E, Commins D, Achim CL, Fox HS, Kolson DL, Grant I, Singer E, Yiannoutsos CT, Sherman S, Gensler G, Moore DJ, Chen T, Soukup VM (2013) Neurovirological correlation with HIV-associated neurocognitive disorders and encephalitis in a HAART-era cohort. J Acquir Immune Defic Syndr 62:487–495
Glass JD, Fedor H, Wesselingh SL, McArthur JC (1995) Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol 38:755–762
Gonzalez R, Cherner M (2008) Co-factors in HIV neurobehavioural disturbances: substance abuse, hepatitis C and aging. Int Rev Psychiatry 20:49–60
Gonzalez E, Dhanda R, Bamshad M, Mummidi S, Geevarghese R, Catano G, Anderson SA, Walter EA, Stephan KT, Hammer MF, Mangano A, Sen L, Clark RA, Ahuja SS, Dolan MJ, Ahuja SK (2001) Global survey of genetic variation in CCR5, RANTES, and MIP-1alpha: impact on the epidemiology of the HIV-1 pandemic. Proc Natl Acad Sci U S A 98:5199–5204
Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK (2005) The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307:1434–1440
Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL (2005) Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS 19:407–411
Gupta S, Bousman CA, Chana G, Cherner M, Heaton RK, Deutsch R, Ellis RJ, Grant I, Everall IP (2011) Dopamine receptor D3 genetic polymorphism (rs6280TC) is associated with rates of cognitive impairment in methamphetamine-dependent men with HIV: preliminary findings. J Neurovirol 17(3):239–247
Heaton RK, Marcotte TD, Mindt MR, Sadek J, Moore DJ, Bentley H, Mccutchan JA, Reicks C, Grant I (2004) The impact of HIV-associated neuropsychological impairment on everyday functioning. J Int Neuropsychol Soc 10:317–331
Heaton RK, Franklin DR, Ellis RJ, Mccutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, GAMST A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, CHARTER Group, HNRC Group (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17(1):3–16
Hinkin CH, Castellon SA, Levine AJ, Barclay TR, Singer EJ (2008) Neurocognition in individuals co-infected with HIV and hepatitis C. J Addict Dis 27:11–17
Kallianpur AR, Levine AJ (2014) Host genetic factors predisposing to HIV-associated neurocognitive disorder. Curr HIV/AIDS Rep 11(3):336–352
Kaul M, Lipton SA (2005) Experimental and potential future therapeutic approaches for HIV-1 associated dementia targeting receptors for chemokines, glutamate and erythropoietin. Neurotox Res 8:167–186
Kelder W, Mcarthur JC, Nance-Sproson T, Mcclernon D, Griffin DE (1998) Beta-chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol 44:831–835
Kraft-Terry SD, Buch SJ, Fox HS, Gendelman HE (2009) A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron 64:133–145
Kumar AM, Fernandez J, Singer EJ, Commins D, Waldrop-Valverde D, Ownby RL, Kumar M (2009) Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol 15:257–274, 1–18
Kumar AM, Ownby RL, Waldrop-Valverde D, Fernandez B, Kumar M (2011) Human immunodeficiency virus infection in the CNS and decreased dopamine availability: relationship with neuropsychological performance. J Neurovirol 17:26–40
Lamers SL, Poon AF, Mcgrath MS (2011) HIV-1 nef protein structures associated with brain infection and dementia pathogenesis. PLoS One 6:e16659
Lane BR, King SR, Bock PJ, Strieter RM, COFFEY MJ, Markovitz DM (2003) The C-X-C chemokine IP-10 stimulates HIV-1 replication. Virology 307:122–134
Lehmann MH, Masanetz S, Kramer S, Erfle V (2006) HIV-1 Nef upregulates CCL2/MCP-1 expression in astrocytes in a myristoylation- and calmodulin-dependent manner. J Cell Sci 119:4520–4530
Letendre S (2011) Central nervous system complications in HIV disease: HIV-associated neurocognitive disorder. Top Antivir Med 19:137–142
Letendre SL, Ellis RJ, Ances BM, Mccutchan JA (2010) Neurologic complications of HIV disease and their treatment. Top HIV Med 18:45–55
Letendre SL, Zheng JC, Kaul M, Yiannoutsos CT, Ellis RJ, Taylor MJ, Marquie-Beck J, Navia B (2011) Chemokines in cerebrospinal fluid correlate with cerebral metabolite patterns in HIV-infected individuals. J Neurovirol 17:63–69
Levine AJ, Singer EJ, Sinsheimer JS, Hinkin CH, Papp J, Dandekar S, Giovanelli A, Shapshak P (2009) CCL3 genotype and current depression increase risk of HIV-associated dementia. Neurobehav HIV Med 1:1–7
Levine AJ, Sinsheimer JS, Bilder R, Shapshak P, Singer EJ (2012) Functional polymorphisms in dopamine-related genes: effect on neurocognitive functioning in HIV+ adults. J Clin Exp Neuropsychol 34:78–91
Levine AJ, Panos SE, Horvath S (2014a) Genetic, transcriptomic, and epigenetic studies of HIV-associated neurocognitive disorder. J Acquir Immune Defic Syndr 65:481–503
Levine AJ, Reynolds S, Cox C, Miller EN, Sinsheimer JS, Becker JT, Martin E, Sacktor N, Neuropsychology Working Group of the Multicenter, A. C. S (2014b) The longitudinal and interactive effects of HIV status, stimulant use, and host genotype upon neurocognitive functioning. J Neurovirol 20:243–257
Lojek E, Bornstein RA (2005) The stability of neurocognitive patterns in HIV infected men: classification considerations. J Clin Exp Neuropsychol 27:665–682
Lu SM, Tremblay ME, King IL, Qi J, Reynolds HM, Marker DF, Varrone JJ, Majewska AK, Dewhurst S, Gelbard HA (2011) HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One 6:e23915
Martin-Thormeyer EM, Paul RH (2009) Drug abuse and hepatitis C infection as comorbid features of HIV associated neurocognitive disorder: neurocognitive and neuroimaging features. Neuropsychol Rev 19:215–231
Masliah E, Achim CL, Ge N, Deteresa R, Terry RD, Wiley CA (1992) Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol 32:321–329
Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, Mccutchan JA, Nelson JA, Atkinson JH, Grant I (1997) Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol 42:963–972
McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, Graham NM, McArthur JH, Selnes OA, Jacobson LP et al (1993) Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology 43:2245–2252
McArthur JC, Brew BJ, Nath A (2005) Neurological complications of HIV infection. Lancet Neurol 4:543–555
Mirowska-Guzel D, Gromadzka G, Mach A, Czlonkowski A, Czlonkowska A (2011) Association of IL1A, IL1B, ILRN, IL6, IL10 and TNF-alpha polymorphisms with risk and clinical course of multiple sclerosis in a Polish population. J Neuroimmunol 236:87–92
Modi WS, Lautenberger J, An P, Scott K, Goedert JJ, Kirk GD, Buchbinder S, Phair J, Donfield S, O'Brien SJ, Winkler C (2006) Genetic variation in the CCL18-CCL3-CCL4 chemokine gene cluster influences HIV Type 1 transmission and AIDS disease progression. Am J Hum Genet 79:120–128
Moore DJ, Masliah E, Rippeth JD, Gonzalez R, Carey CL, Cherner M, Ellis RJ, Achim CL, Marcotte RD, Heaton RK, Grant I (2006) Cortical and subcortical neurodegeneration is associated with HIV neurocognitive impairment. AIDS 20:879–887
Mortimer JA, Snowdon DA, Markesbery WR (2009) The effect of APOE-epsilon4 on dementia is mediated by Alzheimer neuropathology. Alzheimer Dis Assoc Disord 23:152–157
Nolting T, Lindecke A, Koutsilieri E, Maschke M, Husstedt IW, Sopper S, Stuve O, Hartung HP, Arendt G, COMPETENCE NETWORK, H. A (2009) Measurement of soluble inflammatory mediators in cerebrospinal fluid of human immunodeficiency virus-positive patients at distinct stages of infection by solid-phase protein array. J Neurovirol 15:390–400
Pemberton LA, Stone E, Price P, van Bockxmeer F, Brew BJ (2008) The relationship between ApoE, TNFA, IL1a, IL1b and IL12b genes and HIV-1-associated dementia. HIV Med 9:677–680
Persidsky Y, Gendelman HE (2003) Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J Leukoc Biol 74:691–701
Price P, James I, Fernandez S, French MA (2004) Alleles of the gene encoding IL-1alpha may predict control of plasma viraemia in HIV-1 patients on highly active antiretroviral therapy. AIDS 18:1495–1501
Rempel HC, Pulliam L (2005) HIV-1 Tat inhibits neprilysin and elevates amyloid beta. AIDS 19:127–135
Schmidtmayerova H, Nottet HS, Nuovo G, Raabe T, Flanagan CR, Dubrovsky L, Gendelman HE, Cerami A, Bukrinsky M, Sherry B (1996) Human immunodeficiency virus type 1 infection alters chemokine beta peptide expression in human monocytes: implications for recruitment of leukocytes into brain and lymph nodes. Proc Natl Acad Sci U S A 93:700–704
Shulman JM, Chibnik LB, Aubin C, Schneider JA, Bennett DA, De Jager PL (2010) Intermediate phenotypes identify divergent pathways to Alzheimer’s disease. PLoS One 5:e11244
Soontornniyomkij V, Moore DJ, Gouaux B, Soontornniyomkij B, Tatro ET, Umlauf A, Masliah E, Levine AJ, Singer EJ, Vinters HV, Gelman BB, Morgello S, Cherner M, Grant I, Achim CL (2012) Cerebral beta-amyloid deposition predicts HIV-associated neurocognitive disorders in APOE epsilon4 carriers. AIDS 26:2327–2335
Vitek MP, Brown CM, Colton CA (2009) APOE genotype-specific differences in the innate immune response. Neurobiol Aging 30:1350–1360
Weiss JM, Nath A, Major EO, Berman JW (1999) HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol 163:2953–2959
Woods SP, Rippeth JD, Frol AB, Levy JK, Ryan E, Soukup VM, Hinkin CH, Lazzaretto D, Cherner M, Marcotte TD, Gelman BB, Morgello S, Singer EJ, Grant I, Heaton RK (2004) Interrater reliability of clinical ratings and neurocognitive diagnoses in HIV. J Clin Exp Neuropsychol 26:759–778
Zheng J, Thylin MR, Persidsky Y, Williams CE, Cotter RL, Zink W, Ryan L, Ghorpade A, Lewis K, Gendelman HE (2001) HIV-1 infected immune competent mononuclear phagocytes influence the pathways to neuronal demise. Neurotox Res 3:461–484
Acknowledgments
We thank the participants of the National NeuroAIDS Tissue Consortium for their dedication to the advancement of medical treatment for NeuroAIDS. This research was funded primarily by National Institute of Mental Health grant R01-MH096648. Data were also provided by the National Neurological AIDS Bank (U24-MH100929, U01-MH083500, and R24-NS38841) and the California NeuroAIDS Tissue Network (U24-MH100928 and R24-MH59745).
Author contributions
Andrew J. Levine helped conceive of the study, obtain funding, analyze data, and write the manuscript.
Virawudh Soontornniyomkij helped with the study design, carried out immunohistochemistry, and helped write the manuscript.
Cristian L. Achim helped with the study design, helped to carry out immunohistochemistry, and helped write the manuscript.
Eliezer Masliah helped with the study design and carried out MAP2 and SYP immunohistochemistry.
Benjamin B. Gelman carried out brain HIV RNA viral load assays.
Janet S. Sinsheimer helped with the study design, statistical analysis, and in writing the manuscript.
Elyse J. Singer helped with the study design, procuring samples from the NNAB, and writing the manuscript.
David J. Moore helped conceive the study, obtain funding, and write the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 47 kb)
Rights and permissions
About this article

Cite this article
Levine, A.J., Soontornniyomkij, V., Achim, C.L. et al. Multilevel analysis of neuropathogenesis of neurocognitive impairment in HIV. J. Neurovirol. 22, 431–441 (2016). https://doi.org/10.1007/s13365-015-0410-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-015-0410-7