
Abstract
We screened for viral DNA in cerebrospinal fluid samples using next-generation sequencing (NGS) technology to diagnose CNS viral infections. We collected CSF samples from four cases with clinically suspected viral meningoencephalitis. DNA extracted from the samples was analyzed with NGS, and the results were further validated using PCR. Herpes simplex virus 1 (HSV-1) was detected in the CSF of two patients, HSV-2 and human herpes virus type 3 (HHV-3, VZV) in the CSF of two other patients separately. The number of unique reads of the identified viral genes ranged from 144 to 44205 (93.51 to 99.57 %). The coverage of identified viral genes ranged from 12 to 98 % with a depth value of 1.1 to 35, respectively. The results were further confirmed using PCR in three cases. The clinical presentation and outcomes of these four cases were consistent with the diagnostic results of NGS. NGS of CSF samples can be used as a diagnostic assay for CNS viral infection. Its further application for “pan-viral” or even “pan-microbial” screening of CSF might influence the diagnosis of CNS infectious diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The etiology of acute encephalitis remains unclear in more than 50 % of cases despite extensive testing for infectious pathogens in clinical samples including cerebrospinal fluid (CSF) (Glaser et al. 2006). Timely diagnosis is usually hindered by the lack of assays for rapid screening. In recent years, unbiased next-generation sequencing (NGS) has been used in medical microbiology as an emerging and powerful technique due to its low cost and rapid turnaround time. NGS is a novel approach to DNA/RNA sequencing. The ability to generate millions to billions of DNA/RNA sequences per run enables metagenomic analysis and is a significant advantage over traditional Sanger sequencing. NGS is an unbiased assay as it can amplify and sequence the entire DNA content of a sample without using any primers or probes. Theoretically, unbiased NGS facilitates identification of all the potential pathogens in a single assay. NGS technology for comprehensive detection of pathogens contributes to the early diagnosis of infectious diseases (Naccache et al. 2014). Wilson et al. (2014) reported that unbiased NGS identified Leptospira santarosai in the CSF of a patient with severe combined immunodeficiency, which provided a clinical diagnosis and facilitated the use of targeted and efficacious antimicrobial therapy (Wilson et al. 2014). However, no further studies have been reported in the application of NGS for pathogen detection from CSF samples.
Here, we report the development of an efficient, accurate, and comprehensive method based on NGS for the rapid detection and identification of virus directly in CSF specimens, in the context of meningoencephalitis of unknown etiology.
Materials and methods
Sample collection and information
The CSF was collected from the Department of Neurology in Peking Union Medical College Hospital according to standard procedures, snap-frozen, and stored at −20 °C. The patients had signed informed consent, and samples were used for research only. The Institutional Review Board of Peking Union Medical College Hospital and BGI-Shenzhen approved this study. The Ethics Committee of PUMCH approved the use of human subjects for this study. All the patients provided written (signed) informed consent to participate in this study.
DNA extraction, library preparation, and sequencing
DNA was extracted directly from the clinical samples with TIANamp Micro DNA Kit (DP316, TIANGEN BIOTECH). The extracted DNA was performed using a Sigma-Aldrich WGA4 Kit for whole genomic amplification (WGA), subsequently purified the products with QIA quick PCR purification kit (Qiagen, cat no. 28106), and sonicated to a size of 200–300 bp (Bioruptor Pico protocols). The DNA libraries were constructed through end repair, supplemented with adapter overnight followed by PCR amplification, template preparation in the OneTouch system, and sequencing using the BGISEQ-100 platform after quality control (Jeon et al. 2014).
Data treatment and analysis
High-quality sequencing data were generated by removing low-quality reads, adapter contamination, and duplicated reads and discarding those shorter than 35 bp. Human sequence data were excluded and mapped to a human reference (hg19) using Burrows-Wheeler Alignment (BWA), a powerful alignment tool, which is also appropriate for the proton platform (Li and Durbin 2010). After removing human sequences, the remaining sequencing data were aligned to the bacterial, virus, fungal, and protozoan databases. The mapped data were processed for advanced data analysis.
We downloaded the latest version of the microbial reference genomes, from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/).
Currently, our databases contain 680 bacterial genera, 110 viral species related to human diseases, and 54 fungal species that cause human infections. We used the SoapCoverage software from the SOAP website (http://soap.genomics.org.cn/) to calculate the depth and coverage of each species.
PCR and Sanger validation
We carried out sequence-specific PCR identification of herpes simplex virus 1 (HSV-1), HSV-2, and human herpes virus-3 (HHV-3) with a target fragment validation of the NGS results. The specific primers used for the gene amplification are as follows:
-
HSV-1
-
HSV-1-F 5′-GCCAGCGAGACGCTGATGAAG-3′ and
-
HSV-1-R 5′-ACGCAGGTACTCGTGGTG-3′;
-
HSV-2
-
HSV-2-F 5′-CATCGCGTATCACGGCATG-3′ and
-
HSV-2-R 5′-GCTGAATGTGGTAAACACGCT-3′.
The PCR products were analyzed using agarose gel electrophoresis and sequenced with 3730 XL (ABI).
Results
Patient demographics
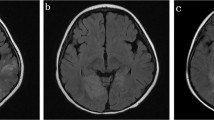
The case series included three male and one female adult patients. All patients were previously healthy and presented with headache and fever with acute onset. Three of them showed behavioral changes and decreased level of consciousness. One patient developed epilepsy. In terms of clinical severity, the modified Rankin Score ranged from 2 to 5 and one case required ICU management. Meningitis was seen in three patients. Lumbar puncture revealed increased opening pressure of CSF (215–330 mmH2O) in all patients. CSF white cell count was elevated in all cases, ranging from 32 to 492 × 106/L. The CSF cytology indicated lymphocytic inflammation. All cases showed mild elevation in CSF protein levels. Two patients had abnormal cerebral parenchyma on brain MRI: one in the bilateral medial temporal lobe and one in the splenium of corpus callosum. All patients received acyclovir intravenously for 3–4 weeks resulting insubstantial recovery. The modified Rankin Score (mRS) was 0–1 at the time of patient’s discharge (Tables 1 and 2 and Fig. 1).

MRI of case no. 1: Abnormal signals were shown in bilateral mesial temporal lobes (arrow) on axial T2 (a), FLAIR (b), and contrast-enhanced T1 (c)
NGS
HSV-1 DNA was detected in the CSF of two patients (case nos. 1 and 3), HSV-2 in the CSF of one patient (case no. 3), and HHV-3 in the CSF of another patient (case no. 4). The number of unique reads of the identified viral gene ranged from 144 to 44205 (93.51–99.57 %). Mapping of the detected reads to the viral genome resulted in a coverage ranging from 12 to 98 % with a depth of 1.1 to 35, respectively. The number of unique reads, percentage, coverage, and depth of the identified viral DNA sequences are listed in Table 3 and Fig. 2.

NGS of virus in patients’ CSF. a In case no. 1, the majority of viral reads (44,205 of 44,403 reads; 99.55 %) corresponded to HSV-1, with a coverage of 98 %. b In case no. 2, the majority of viral reads (2272 of 2295 reads; 99.00 %) corresponded to HSV-1, with a coverage of 53 %. c In case no. 3, the majority of viral reads (464 of 466 reads; 99.57 %) corresponded to HSV-2, with a coverage of 27 %. d In case no. 4, the majority of viral reads (144 of 154 reads; 93.51 %) corresponded to VZV(HHV-3) with a coverage of 12 %
We validated the NGS results using PCR analysis and Sanger sequencing. Specific primers were designed for the HSV-1 and HSV-2 sequences, and PCR was carried out for the case no. 1, no. 2, and no. 3. The CSF sample of the case no. 4 was not available after NGS. No PCR analysis of case 4 was performed. The results showed that the amplicon was consistent with our expectation, and the read from Sanger sequencing was consistent with HSV-1 and HSV-2 genome, respectively (Fig. 3).

PCR amplification of HSV-1 and HSV-2 was followed by agarose gel electrophoresis to confirm HSV-1 and HSV-2 sequences for case no. 1 (a), no. 2 (b), and no. 3 (c) respectively. M DNA markers of DL2000 or Trans 2K Plus, N negative control. The numbers 171, 105, and 522 represent the sample codes
Discussion
Conventional CSF tests, especially for CNS viral infection, have limited sensitivity given the pathogen diversity and lack of standardized assays. Approximately 60 % of cases with meningoencephalitis remain undiagnosed, despite extensive clinical laboratory testing (Glaser et al. 2006). Etiological diagnosis based on culture results, serologic findings, and pathogen-specific PCR assays are inaccurate since more than 100 different infectious agents cause encephalitis. NGS is a potentially revolutionary screening modality in the identification of pathogens, including rare and newly identified viruses. The use of NGS technology for comprehensive detection of pathogens from CSF samples is of great interest since the first reported case in 2014 (Wilson et al. 2014). Our study aimed at exploring the feasibility of using NGS of CSF in CNS viral infection.
NGS offers the possibility of pathogen identification without a prior knowledge of the target. Theoretically, given sufficiently long read lengths, multiple hits to the microbial genome, and a well-annotated reference database, nearly all microorganisms can be uniquely identified on the basis of specific nucleic acid sequences. In our study, a large number of unique reads of neurotropic viral genome were detected in the patient’s CSF. These viral reads were absent in the control samples of three unrelated patients analyzed concomitantly. The NGS results were confirmed by PCR with Sanger methods. The clinical presentation, the CSF, and neuroimaging data as well as the response to antiviral treatment were consistent with the diagnosis of viral infection detected by NGS. The coverage of VZV (12 %) in case no. 4 was relatively low compared with the other cases. However, the majority of viral reads (93.51 %, 144/154) corresponded to VZV and all the mapped reads were distributed uniformly in the whole genome region of HZV. According to the published report by Wilson et al. 2014, the unique reads mapped to Leptospira santarosai with a coverage of 3.7 and 2.2 % for chromosomes 1 and 2, respectively, which are lower than our values. Integrating with the number of unique reads, dispersive distribution of reads along the genome, we consider our results reliable in spite of the differences in coverage.
NGS is a time-saving and accurate approach for the molecular diagnosis of diseases. Compared with traditional clinical diagnosis, NGS techniques dramatically reduced the diagnostic period to less than 3 days. Further, NGS technology is highly accurate and specific in diagnosis. The availability of genome sequences using NGS has revolutionized the field of infectious diseases. Indeed, more than 38,000 bacterial and 5000 viral genomes have been sequenced to date, including representatives of all significant human pathogens (Fournier et al. 2014). The copious data have not only advanced fundamental research but also have implications for clinical microbiology in the screening for pathogens, virulence factors, or antibiotic resistance (Sherry et al. 2013; Padmanabhan et al. 2013). NGS technologies are expected to have a major impact on routine clinical and medical diagnostics in the near future due to the increasing availability of a multitude of platforms and dramatically decreased costs of sequencing.
This study highlights the feasibility of using NGS of CSF as a diagnostic tool for CNS viral infection. Though it remains largely unexplored, its routine application in the near future for “pan-viral” or even “pan-microbial” screening of CSF might alter the clinical diagnosis of CNS infectious diseases.
Abbreviations
- CSF:
-
Cerebrospinal fluid
- NGS:
-
Next-generation sequencing
- EEG:
-
Electroencephalogram
- HSV:
-
Human herpes virus
- VZV:
-
Varicella zoster virus
References
Fournier PE, Dubourg G, Raoult D (2014) Clinical detection and characterization of bacterial pathogens in the genomics era. Genome Med 6(11):114. doi:10.1186/s13073-014-0114-2. eCollection 2014
Glaser CA, Honarmand S, Anderson LJ, Schnurr DP, Forghani B, Cossen CK, Schuster FL, Christie LJ, Tureen JH (2006) Beyond viruses: clinical profiles and etiologies associated with encephalitis. Clin Infect Dis 43:1565–1577
Jeon YJ, Zhou Y, Li Y, et al. (2014) The feasibility study of non-invasive fetal trisomy 18 and 21 detection with semiconductor sequencing platform. PLoSOne.2014;20;9(10):e110240. doi: 10.1371/journal.pone.0110240. eCollection 2014
Li H, Durbin R (2010) Fast and accurate short read alignment with Burrows Wheeler transform. Bioinformatics 26(5):589–595
Naccache SN, Federman S, Veeraraghavan N, Zaharia M, Lee D, Samayoa E, Bouquet J, Greninger AL, Luk KC, Enge B, Wadford DA, Messenger SL, Genrich GL, Pellegrino K, Grard G, Leroy E, Schneider BS, Fair JN, Martínez MA, Isa P, Crump JA, DeRisi JL, Sittler T, Hackett J Jr, Miller S, Chiu CY (2014) A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res 24:1180–1192
Padmanabhan R, Mishra AK, Raoult D, Fournier PE (2013) Genomics and metagenomics in medical microbiology. J Microbiol Methods 95(3):415–424
Sherry NL, Porter JL, Seemann T, Watkins A, Stinear TP, Howden BP (2013) Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J Clin Microbiol 51(5):1396–1401
Wilson MR, Naccache SN, Samayoa E, Biagtan M, Bashir H, Yu G, Salamat SM, Somasekar S, Federman S, Miller S, Sokolic R, Garabedian E, Candotti F, Buckley RH, Reed KD, Meyer TL, Seroogy CM, Galloway R, Henderson SL, Gern JE, DeRisi JL, Chiu CY (2014) Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med 370:2408–2417
Authors’ contributions
Hongzhi Guan, MD, is the principal investigator who drafted the original manuscript. Ao Shen, PhD, participated in laboratory analysis of CSF and drafted parts of the original manuscript. Xia Lv, MD, was involved in case and sample collection as well as analysis or interpretation of data. Xunzhe Yang, MD, was involved in case and sample collection, analysis, or interpretation of the data. Haitao Ren, BA, Yanhuan Zhao, BA, Yinxin Zhang, PhD, and Yanping Gong, PhD, participated in the laboratory analysis of CSF. Peixiang Ni, PhD, analyzed or interpreted the data. Honglong Wu, PhD, Yicheng Zhu, MD, and Liying Cui, MD, conceptualized the study and revised the manuscript.
Conflict of interest
We declare that we have no financial and personal relationships with other people or organizations that may inappropriately influence our work. There is no professional or other personal interest of any nature or kind in any product, service, and/or company that could be construed as influencing the position presented in or the review of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Hongzhi Guan and Ao Shen contributed equally to this work.
Rights and permissions
About this article

Cite this article
Guan, H., Shen, A., Lv, X. et al. Detection of virus in CSF from the cases with meningoencephalitis by next-generation sequencing. J. Neurovirol. 22, 240–245 (2016). https://doi.org/10.1007/s13365-015-0390-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-015-0390-7