
Abstract
Combination antiretroviral therapy (ART) has altered the outcomes of HIV infection in treated populations by greatly reducing the incidence of opportunistic infections, cancer, and HIV-associated dementia. Despite these benefits, treated patients remain at high risk of chronic diseases affecting the peripheral organs and brain. Generally, these morbidities are attributed to persistence of latent HIV in resting T cells, chronic inflammation, and metabolic effects of ART. This review makes the case that monocytes/macrophages warrant attention as persistent reservoirs of HIV under ART, source of systemic and brain inflammation, and important targets for HIV eradication to control chronic HIV diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
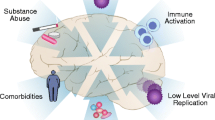
The morbidity and mortality of HIV infection have changed significantly in countries with access to antiretroviral therapy (ART). HIV infection can now be managed as a chronic condition with continued treatment resulting in stable suppression of virus replication and immunological improvement. Because individuals on successful ART generally do not develop immunodeficiency, the previously fatal complications of opportunistic infections and cancers can be largely prevented. In the central nervous system (CNS), ART dramatically reduced the prevalence of HIV dementia, the most severe form of HIV-associated neurologic diseases (HAND) (Antinori et al. 2007) and a major AIDS-defining disease in the pre-ART period (Navia et al. 1986). Despite these benefits, treated patients remain at high risk for chronic diseases affecting the circulatory system, gut, lung, lipid, bone, and energy metabolism. Some of these abnormalities appear to reflect a state of accelerated aging. In addition, a large fraction of HIV patients on ART, an estimated 50 %, exhibit milder, sub-dementia forms of HIV-associated brain disease known as asymptomatic neurocognitive impairment (ANI) and mild neurocognitive disorder (MND) (Antinori et al. 2007; Harezlak et al. 2011; Heaton et al. 2010). ANI and MND, rather than dementia, now constitute the majority of HIV neurological cases diagnosed in people on ART with sustained plasma virus suppression and improved or normal CD4 T cell levels (Antinori et al. 2007; Harezlak et al. 2011; Heaton et al. 2010). The key questions in HIV research are therefore the following: what are the virological and host mechanisms responsible for these morbidities, and how can these illnesses be prevented, treated, or cured (the latter likely requiring elimination of virus-infected cellular reservoirs)?
The current view attributes chronic diseases observed in individuals on ART to chronic inflammation caused by residual HIV in tissues, with additional complications of metabolic effects of prolonged antiviral drug administration (Lake and Currier 2013; Sandler and Douek 2012). While ART prevents HIV replication (infection of new cells), a large fraction of patients on effective therapy still have low (several copies/ml) but persistent levels of free virus in plasma which is detectable by sensitive PCR and culture methods (Palmer et al. 2008; Siliciano et al. 2003). This “residual” plasma viremia is stable, insensitive to antiretroviral therapy, and it presents the major obstacle to HIV cure (reviewed in (Katlama et al. 2013; Shan and Siliciano 2013)). The origins of this virus are not fully known, but one conspicuous cellular niche for HIV persistence is the pool of resting memory CD4+ T cells carrying latent HIV (reviewed in (Shan and Siliciano 2013)). In the absence of HIV expression, these cells do not undergo viral cytolysis and are not recognized by virus specific cytotoxic T cells, but they can produce virus and spread new infection upon stimulation (Ho et al. 2013). Importantly, latently infected T cells were shown to decay at slow rates, with estimated t (1/2) of 44 months, consistent with persistence and stability of residual HIV in plasma (Shan and Siliciano 2013; Siliciano et al. 2003). Extensive efforts are being devoted to testing strategies to induce and eradicate latent HIV in T cells, including by the novel methods of somatic cell gene editing (Hu et al. 2014; Shan and Siliciano 2013). However, T cell reservoirs may not fully account for residual HIV in treated patients. Careful genetic analysis of virus in patients on therapy (Bailey et al. 2006; Palmer et al. 2008) or after discontinuation of therapy (Chun et al. 2000) showed that, in the majority of subjects, residual and rebounded HIV in plasma, respectively, were genetically distinct from virus present in pre-existing T cell reservoirs, suggesting that these cells are not the sole source of residual viremia. Notably, attempts to reactivate latent virus in T cells in vivo of patients on ART with immunological stimulators have been thus far unsuccessful (Mitsuyasu et al. 2007). HIV reactivation in T cells can be achieved using chromatin modifying agents such as valproic acid or vorinostat (Del Prete et al. 2014; Elliott et al. 2014; Lehrman et al. 2005; Wei et al. 2014). However, evidence that increased HIV transcription in T cells caused by these agents may facilitate HIV cure is mixed. Earlier clinical trials suggested depletion of latent HIV infection in T cells after treatment with valproic acid (Lehrman et al. 2005), whereas recent study with larger group of patients showed no effects of a potent HDAC inhibitor vorinostat on plasma virus levels, frequency of latently infected cells, and frequency of HIV specific T cells (Elliott et al. 2014). Because residual HIV in individuals on effective suppressive ART shows only limited genetic diversification overtime, with no new appearance of ART resistance mutations (Bailey et al. 2006; Wong et al. 1997), it is unlikely that this virus arises from active virus replication in an unidentified tissue. In some studies, however, residual viremia can be reduced (but not eliminated) upon intensification of treatment (Baroncelli et al. 2015). Together, these considerations suggest that other persistently infected cells, in addition to memory T cells, contribute to stable residual viremia and chronic inflammatory diseases in patients on effective ART.
HIV-infected macrophages are one such cell type worth consideration, and several excellent reviews have recently elaborated on this subject (Costiniuk and Jenabian 2014; Dey et al. 2012; Stevenson 2014; Tan and Sattentau 2013; Watters et al. 2013). Infected macrophages have been found at low but measurable frequencies in the lung and duodenal tissue of patients on ART with undetectable plasma virus (Cribbs et al. 2015; Zalar et al. 2010) and in the brain tissues of SIV-infected macaques on ART (Clements et al. 2002). The potential role of macrophages as a lasting HIV sanctuary is further supported by HIV biology in these cells. Macrophages are natural target cells for lentiviruses including HIV. Although they are terminally differentiated, non-dividing cells, lentiviruses have evolved the mechanisms to transport viral core to the intact nucleus, integrate viral DNA into transcriptionally active sites on chromosomes, and initiate chronic productive infection in these cells. Macrophages support HIV infection without undergoing viral cytolysis or apoptosis, and infected cells are insensitive to the currently available antiretroviral drugs and antiviral CTL; as a consequence, infected macrophages have long life span compared to productively infected T cells (Borjabad et al. 2011; Busca et al. 2012; Cribbs et al. 2015; Le Douce et al. 2010; Murphy et al. 2008; Vojnov et al. 2012). Both HIV and SIV have evolved strategies to overcome macrophage restriction factors, contributing to their persistence in these cells (reviewed in (Stevenson 2014)). The ability of HIV to assemble and accumulate in intracellular compartments connected to extracellular space (reviewed in (Tan and Sattentau 2013)) may further protect virus from immunological responses and contribute to virus transmission despite ART (Duncan et al. 2013). At present, it is an open question whether monocytes/macrophages can also support HIV latency similar to memory T cells. Many of the studies on latency were conducted in transformed macrophage cell lines (Kumar et al. 2014); the few published studies of primary cells in patients indicate that at least intestinal and lung macrophages remain productively infected under suppressive ART (Cribbs et al. 2015; Zalar et al. 2010). Transient SIV latency in macrophages/microglia was documented during early stages of infection in macaques (Barber et al. 2006), and one recent report demonstrated presence of unexpressed HIV DNA in macrophages/microglia in autopsy brain tissue from patients who died with presymptomatic HIV infection (Thompson et al. 2011). It should be noted that because HIV infection of macrophages is largely non-lytic and infected cells may be able to avoid host immune responses (Busca et al. 2012; Vojnov et al. 2012), viral latency may not be essential for their survival as HIV reservoirs under ART. It should be of interest to determine whether HIV RNA present in residual-infected macrophages under ART bears sequence similarity with that of HIV RNA of residual HIV in plasma.
Another important consideration regarding role of macrophages as viral reservoirs is the longevity or turnover of infected cells under long-term therapy. In contrast to infected T cells, the dynamics of HIV-infected macrophages is largely unknown (Costiniuk and Jenabian 2014). In one longitudinal study in a small number of patients in Thailand, a subset of subjects maintained HIV DNA positive CD14+ monocytes in the blood for 3.5 years of follow-up since initiation of treatment while CD14+ depleted lymphocytes were HIV negative during last year of follow-up (Shiramizu et al. 2012). Tissue macrophages may have longer life spans than circulating monocytoid cells (Murphy et al. 2008). The half-life of human perivascular macrophages has been estimated in months to years, whereas microglia turnover move slowly (Kofler and Wiley 2011). In the setting of disease, microglia turnover may be faster and approximate than of perivascular macrophages (Kofler and Wiley 2011). In non-human primates, monocytes turn over more rapidly in response to viral infection and are replaced at a higher rate from the bone marrow (Burdo et al. 2010). It is conceivable that the increased turnover observed in the blood actually involves the invasion of bone marrow-derived monocytes into tissues. This scenario is supported by bioinformatics analysis demonstrating the similarity of HIV sequences in the brain, blood, and bone marrow, suggesting a directionality of virus trafficking (Liu et al. 2000). It is conceivable that HIV-infected macrophages and microglia could be replaced by uninfected cells with long-term antiretroviral therapy. However, even with the limited information on macrophage longevity (Cribbs et al. 2015; Shiramizu et al. 2012; Zalar et al. 2010), strategies to target infected macrophages for HIV eradication should also be considered.
While macrophages may serve as an important source of residual virus under ART, the subversion of the physiological functions of these cells by HIV infection, combined with longevity of infected macrophages, play important roles in HIV pathogenesis at many levels of human physiology (Assimakopoulos et al. 2014; Brown 2015; Burdo et al. 2013; Cavarelli and Scarlatti 2014). Paradoxically, the widespread use of ART, through diminution of CD4 T cell depletion, has exposed the core lentiviral nature of HIV as a pathogen adapted to survive and cause slow progressive disease, including in the nervous system, through colonization of macrophages. The role of macrophages and microglia in HIV-associated neurologic diseases (HAND) has been known for many years. HIV enters the CNS early after primary infection (Valcour et al. 2012), likely by migration of infected monocytes (Saini and Potash 2014; Valcour et al. 2013) through blood-brain barrier (Persidsky et al. 2000). Subsequently, HIV persists in the brain in microglia and perivascular macrophages, and to a lesser extent astrocytes, and the process of viral neuropathogenesis is believed to be mediated by inflammatory and cytotoxic products secreted by these cells (Lipton and Gendelman 1995). This is particularly evident in the case of HIV/SIV encephalitis where the levels of neuronal damage correlate with HIV brain burdens (Budka 2005). However, even in the pre-ART era, it was clear that high levels of HIV replication in the brain are not required for sustaining HIV brain disease. In many patients, dementia correlates better with activated macrophages/microglia than productive virus infection in the brain (Glass et al. 1995). Immune activation of macrophages/microglia and astrocytes, although to a much more modest degree, can also be observed in patients on ART with MND (Tavazzi et al. 2014). The HIV brain burdens in these patients are often below level of detection (Gelman et al. 2013). These results suggest that MND and HAD may represent a disease continuum linked by the common mechanism of glial cell activation. This mechanism may include expression of viral and cellular inflammatory products by chronically infected glial cells, with the possibility that pharmacologic regimens including antiretroviral drugs with high CNS penetration may improve cognitive performance in some (Smurzynski et al. 2011) but not all tested patients (Marra et al. 2009). In support of the role of inflammation, per se, it has been shown that soluble CD14 (sCD14), a soluble form of the monocyte endotoxin receptor present on monocytes and macrophages correlates with CD4 nadir, and both are strong predictors of the severity of neurocognitive impairment (Ellis et al. 2011; Fischer-Smith et al. 2001; Lyons et al. 2011; McCombe et al. 2013). CD4+ T cell nadir is also predictive of opportunistic infections as well as death from all causes in patients on ART (Pulliam et al. 1997; Sandler et al. 2011). This raises the possibility that monocyte/macrophage activation is important not just in the CNS pathologies but in the overall HIV pathogenesis. In fact, because ART neither completely restores immunologic functions nor prevents cognitive deficits in HIV infection, it is conceivable that inflammatory processes initiated in the periphery may affect both immunity and neurocognitive performance.
One of the aspects of the HIV brain disease field that has greatly contributed to our understanding of HIV pathogenesis in general has been the characterization of the role of monocytes/macrophages subsets in disease. Monocytes with a more mature phenotype (CD14+/CD16+) have been shown to be increased in the blood in HIV infection, with even higher frequency of expression in HIV dementia (Pulliam et al. 1997). It is also evident that CD16+ monocytes are decreased with ART treatment, and furthermore, the frequency of CD14+/CD163+/CD16+ monocytes in circulation correlates directly with viral load and inversely with CD4+ T cell count (Fischer-Smith et al. 2008a). The notion that this monocyte subset invades the CNS has been demonstrated by in vitro (Pulliam et al. 1997) and by immunohistochemical studies in human and macaque CNS by Rappaport’s group and others (Fischer-Smith et al. 2001, 2008a; Williams et al. 2001a) as well as in visceral tissues (Tavazzi et al. 2014; Walker et al. 2014; Yearley et al. 2006).
The connection between HIV/SIV infection, macrophages, and immune suppression has also been suggested based on immune polarization. An increase in M2-“like” cells, possibly alternatively activated or regulatory macrophages (Caescu et al. 2015; Murray et al. 2014), could be responsible for both CNS disease, other comorbid conditions in AIDS, as well as immune dysfunction. Macrophage colony-stimulating factor (M-CSF) expression is increased in HIV infection, likely driving production on monocytes from the bone marrow and differentiation toward M2. M2 macrophages (as generated in response to M-CSF) are preferentially infected by HIV (Kalter et al. 1991); this may have profound consequences for increasing macrophage targets for infection, and at the same time, polarization immune responses in toward immune suppression.
An additional intersecting pathway between peripheral and brain manifestations of HIV infection is chronic interferon (IFN) stimulation and the consequent dysregulation of interferon-stimulated genes (Borjabad et al. 2011; Gelman et al. 2013; Pulliam 2014; Pulliam et al. 2014; Roberts et al. 2004). In fact, the ability to control Type I interferon responses may also explain why sooty mangabeys and African green monkeys do not get AIDS from SIV infection, whereas rhesus macaques are susceptible to disease (Jacquelin et al. 2009), although there are dissenting views (Bosinger et al. 2013). In the CNS, Type I IFN was shown to control HIV and SIV expression in the brain and neuropathogenesis (Clements et al. 2002; He et al. 2014) but also contributed to brain disease in some systems (Sas et al. 2009). We suspect that there is a connection in both peripheral and brain diseases between altered monocyte/macrophage homeostasis, immune polarization, and the interferon response. IFN-α is known to induce the M2 cytokine, IL-10 (Aman et al. 1996), by recruiting IFN regulatory factor 1 and Stat3 (Ziegler-Heitbrock et al. 2003). IL-10, together with M-CSF, has also been demonstrated to promote the development of CD14+/CD16+ monocytes (Li et al. 2005). As IL-10 is an important immunosuppressive Th2/M2 cytokine, the process leading to expansion of CD14+/CD163+/CD16+ monocytes/macrophages likely has profound implications in the development of CNS disease and immunosuppression.
While macrophage/microglial and also astrocytic infection of the CNS represent important obstacles for HIV eradication, altered inflammatory pathways likely provide more direct explanations for both the neurocognitive impairment and immune dysfunction remaining in successfully treated patients. In the setting of ART, despite adequate control of viral replication, inflammatory pathways, including IFN-activated genes, remain activated above basal levels, contributing to the neuro- and immune-pathogenesis. These processes would serve to promote the accumulation of M2 and/or regulatory macrophages in CNS as well as the other organs, as we have observed in patients with HIVE (Tavazzi et al. 2014). In view of the importance of altered monocyte/macrophage homeostasis, trafficking, and immune polarization (Burdo et al. 2010; Fischer-Smith et al. 2008a, b; Fischer et al. 2014; Hasegawa et al. 2009; Williams et al. 2001a, b), there is an urgent need for pharmacologic strategies applied to HIV infection in order to successfully modulate inflammation and immune polarization. Such an effort will require interdisciplinary approaches combining support from relevant NIH grant programs (i.e., NIAID, NIMH, NINDS as well as other institutes) and an evolution in the mind-set of the academic and industry scientists which has up to now been highly successful, but for the most part focused on viral targets for therapeutic intervention. The timely interactions and synergy between AIDS and HAND programs at NIH provides a unique opportunity to address common pathogenic pathways in diverse manifestations of HIV infection under ART.
References
Aman MJ, Tretter T, Eisenbeis I, Bug G, Decker T, Aulitzky WE, Tilg H, Huber C, Peschel C (1996) Interferon-alpha stimulates production of interleukin-10 in activated CD4+ T cells and monocytes. Blood 87:4731–4736
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE (2007) Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69:1789–1799
Assimakopoulos SF, Dimitropoulou D, Marangos M, Gogos CA (2014) Intestinal barrier dysfunction in HIV infection: pathophysiology, clinical implications and potential therapies. Infection 42:951–959
Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, Haggerty CM, Kamireddi AR, Liu Y, Lee J, Persaud D, Gallant JE, Cofrancesco J Jr, Quinn TC, Wilke CO, Ray SC, Siliciano JD, Nettles RE, Siliciano RF (2006) Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J Virol 80:6441–6457
Barber SA, Gama L, Dudaronek JM, Voelker T, Tarwater PM, Clements JE (2006) Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus-macaque model. J Infect Dis 193:963–970
Baroncelli S, Pirillo MF, Galluzzo CM, Antoni AD, Ladisa N, Francisci D, d’Ettorre G, Segala D, Vivarelli A, Sozio F, Cirioni O, Weimer LE, Fragola V, Parruti G, Floridia M (2015) Rate and determinants of residual viremia in multidrug-experienced patients successfully treated with raltegravir-based regimens. AIDS Res Hum Retrovir 31:71–77
Borjabad A, Morgello S, Chao W, Kim S-Y, Brooks AI, Murray J, Potash MJ, Volsky DJ (2011) Significant effects of antiretroviral therapy on global gene expression in brain tissues of patients with HIV-1-associated neurocognitive disorders. PLoS Pathog 7:e1002213
Bosinger SE, Johnson ZP, Folkner KA, Patel N, Hashempour T, Jochems SP, Del Rio Estrada PM, Paiardini M, Lin R, Vanderford TH, Hiscott J, Silvestri G (2013) Intact type I interferon production and IRF7 function in sooty mangabeys. PLoS Pathog 9:e1003597
Brown A (2015) Understanding the MIND phenotype: macrophage/microglia inflammation in neurocognitive disorders related to human immunodeficiency virus infection. Clin Transl Med 4:7
Budka H (2005) The neuropathology of HIV-associated brain disease. In: Gendelman HE, Grant I, Everall IP, Lipton SA, Swindells S (eds) The neurology of AIDS. Oxford University Press, New York, pp 375–391
Burdo TH, Soulas C, Orzechowski K, Button J, Krishnan A, Sugimoto C, Alvarez X, Kuroda MJ, Williams KC (2010) Increased monocyte turnover from bone marrow correlates with severity of SIV encephalitis and CD163 levels in plasma. PLoS Pathog 6:e1000842
Burdo TH, Lackner A, Williams KC (2013) Monocyte/macrophages and their role in HIV neuropathogenesis. Immunol Rev 254:102–113
Busca A, Saxena M, Kumar A (2012) Critical role for antiapoptotic Bcl-xL and Mcl-1 in human macrophage survival and cellular IAP1/2 (cIAP1/2) in resistance to HIV-Vpr-induced apoptosis. J Biol Chem 287:15118–15133
Caescu CI, Guo X, Tesfa L, Bhagat TD, Verma A, Zheng D, Stanley ER (2015). Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood
Cavarelli M, Scarlatti G (2014) HIV-1 infection: the role of the gastrointestinal tract. Am J Reprod Immunol 71:537–542
Chun T-W, Davey RT Jr, Ostrowski M, Shawn Justement J, Engel D, Mullins JI, Fauci AS (2000) Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat Med 6:757–761
Clements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak M Jr, Tarwater PM, Lifson JD, Zink MC (2002) The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis 186:905–913
Costiniuk CT, Jenabian MA (2014) The lungs as anatomical reservoirs of HIV infection. Rev Med Virol 24:35–54
Cribbs SK, Lennox J, Caliendo AM, Brown LA, Guidot DM (2015) Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res Hum Retrovir 31:64–70
Del Prete GQ, Shoemaker R, Oswald K, Lara A, Trubey CM, Fast R, Schneider DK, Kiser R, Coalter V, Wiles A, Wiles R, Freemire B, Keele BF, Estes JD, Quiňones OA, Smedley J, Macallister R, Sanchez RI, Wai JS, Tan CM, Alvord WG, Hazuda DJ, Piatak M Jr, Lifson JD (2014) Effect of suberoylanilide hydroxamic acid (SAHA) administration on the residual virus pool in a model of combination antiretroviral therapy-mediated suppression in SIVmac239-infected indian rhesus macaques. Antimicrob Agents Chemother 58:6790–6806
Dey J, Ditzler S, Knoblaugh SE, Hatton BA, Schelter JM, Cleary MA, Mecham B, Rorke-Adams LB, Olson JM (2012) A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol Cell Biol 32:4104–4115
Duncan CJ, Russell RA, Sattentau QJ (2013) High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. AIDS 27:2201–2206
Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, Brown G, Roney J, Watson J, Prince MH, Hoy JF, Chomont N, Fromentin R, Procopio FA, Zeidan J, Palmer S, Odevall L, Johnstone RW, Martin BP, Sinclair E, Deeks SG, Hazuda DJ, Cameron PU, Sékaly RP, Lewin SR (2014) Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog 10:e1004473
Ellis RJ, Badiee J, Vaida F, Letendre S, Heaton RK, Clifford D, Collier AC, Gelman B, McArthur J, Morgello S, McCutchan JA, Grant I (2011) CD4 nadir is a predictor of HIV neurocognitive impairment in the era of combination antiretroviral therapy. AIDS 25:1747–1751
Fischer T, Wyatt CM, D’Agati VD, Croul S, McCourt L, Morgello S, Rappaport J (2014) Mononuclear phagocyte accumulation in visceral tissue in HIV encephalitis: evidence for increased monocyte/macrophage trafficking and altered differentiation. Curr HIV Res 12:201–212
Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L’Heureux D, Regulier EG, Richardson MW, Amini S, Morgello S, Khalili K, Rappaport J (2001) CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol 7:528–541
Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J (2008a) Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol 14:318–326
Fischer-Smith T, Tedaldi EM, Rappaport J (2008b) CD163/CD16 coexpression by circulating monocytes/macrophages in HIV: potential biomarkers for HIV infection and AIDS progression. AIDS Res Hum Retrovir 24:417–421
Gelman BB, Lisinicchia JG, Morgello S, Masliah E, Commins D, Achim CL, Fox HS, Kolson DL, Grant I, Singer E, Yiannoutsos CT, Sherman S, Gensler G, Moore DJ, Chen T, Soukup VM (2013) Neurovirological correlation with HIV-associated neurocognitive disorders and encephalitis in a HAART-era cohort. J Acquir Immune Defic Syndr 62:487–495
Glass JD, Fedor H, Wesselingh SL, McArthur JC (1995) Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol 38:755–762
Harezlak J, Buchthal S, Taylor M, Schifitto G, Zhong J, Daar E, Alger J, Singer E, Campbell T, Yiannoutsos C, Cohen R, Navia B (2011) Persistence of HIV-associated cognitive impairment, inflammation, and neuronal injury in era of highly active antiretroviral treatment. AIDS 25:625–633
Hasegawa A, Liu H, Ling B, Borda JT, Alvarez X, Sugimoto C, Vinet-Oliphant H, Kim WK, Williams KC, Ribeiro RM, Lackner AA, Veazey RS, Kuroda MJ (2009) The level of monocyte turnover predicts disease progression in the macaque model of AIDS. Blood 114:2917–2925
He H, Sharer LR, Chao W, Gu CJ, Borjabad A, Hadas E, Kelschenbach J, Ichiyama K, Do M, Potash MJ, Volsky DJ (2014) Enhanced human immunodeficiency virus Type 1 expression and neuropathogenesis in knockout mice lacking Type I interferon responses. J Neuropathol Exp Neurol 73:59–71
Heaton RK, Clifford DB, Franklin DR Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I (2010) HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 75:2087–2096
Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF (2013) Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155:540–51
Hu W, Kaminski R, Yang F, Zhang Y, Cosentino L, Li F, Luo B, Alvarez-Carbonell D, Garcia-Mesa Y, Karn J, Mo X, Khalili K (2014) RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc Natl Acad Sci U S A 111:11461–11466
Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, Dillies MA, Roques P, Butor C, Silvestri G, Giavedoni LD, Lebon P, Barre-Sinoussi F, Benecke A, Muller-Trutwin MC (2009) Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest 119:3544–3555
Kalter DC, Nakamura M, Turpin JA, Baca LM, Hoover DL, Dieffenbach C, Ralph P, Gendelman HE, Meltzer MS (1991) Enhanced HIV replication in macrophage colony-stimulating factor-treated monocytes. J Immunol 146:298–306
Katlama C, Deeks SG, Autran B, Martinez-Picado J, van Lunzen J, Rouzioux C, Miller M, Vella S, Schmitz JE, Ahlers J, Richman DD, Sekaly RP (2013) Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet 381:2109–2117
Kofler J, Wiley CA (2011) Microglia: key innate immune cells of the brain. Toxicol Pathol 39:103–114
Kumar A, Abbas W, Herbein G (2014) HIV-1 latency in monocytes/macrophages. Viruses 6:1837–60
Lake JE, Currier JS (2013) Metabolic disease in HIV infection. Lancet Infect Dis 13:964–975
Le Douce V, Herbein G, Rohr O, Schwartz C (2010) Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 7:32
Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, Coffin JM, Bosch RJ, Margolis DM (2005) Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366:549–555
Li G, Kim YJ, Broxmeyer HE (2005) Macrophage colony-stimulating factor drives cord blood monocyte differentiation into IL-10 (high) IL-12absent dendritic cells with tolerogenic potential. J Immunol 174:4706–4717
Lipton S, Gendelman HE (1995) Dementia associated with the acquired immunodeficiency syndrome. N Engl J Med 233:934–940
Liu Q-H, Williams DA, McManus C, Baribaud F, Doms RW, Schols D, De Clercq E, Kotlikoff MI, Collman RG, Freedman BD (2000) HIV-1 gp120 and chemokines activate ion channels in primary macrophages through CCR5 and CXCR4 stimulation. Proc Natl Acad Sci U S A 97:4832–4837
Lyons JL, Uno H, Ancuta P, Kamat A, Moore DJ, Singer EJ, Morgello S, Gabuzda D (2011) Plasma sCD14 is a biomarker associated with impaired neurocognitive test performance in attention and learning domains in HIV infection. J Acquir Immune Defic Syndr 57:371–379
Marra CM, Zhao Y, Clifford DB, Letendre S, Evans S, Henry K, Ellis RJ, Rodriguez B, Coombs RW, Schifitto G, McArthur JC, Robertson K, Team ACTGS (2009) Impact of combination antiretroviral therapy on cerebrospinal fluid HIV RNA and neurocognitive performance. AIDS 23:1359–1366
McCombe JA, Vivithanaporn P, Gill MJ, Power C (2013) Predictors of symptomatic HIV-associated neurocognitive disorders in universal health care. HIV Med 14:99–107
Mitsuyasu R, Gelman R, Cherng DW, Landay A, Fahey J, Reichman R, Erice A, Bucy RP, Kilby JM, Lederman MM, Hamilton CD, Lertora J, White BL, Tebas P, Duliege AM, Pollard RB, Team ACTGS (2007) The virologic, immunologic, and clinical effects of interleukin 2 with potent antiretroviral therapy in patients with moderately advanced human immunodeficiency virus infection: a randomized controlled clinical trial–AIDS clinical trials group 32. Arch Intern Med 167:597–605
Murphy J, Summer R, Wilson AA, Kotton DN, Fine A (2008) The prolonged life-span of alveolar macrophages. Am J Respir Cell Mol Biol 38:380–385
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA (2014) Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41:14–20
Navia BA, Jordan BD, Price RW (1986) The AIDS dementia complex: I. Clinical features. Ann Neurol 19:517–524
Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, Kempf DJ, Mellors JW, Coffin JM, King MS (2008) Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 105:3879–3884
Persidsky Y, Zheng J, Miller D, Gendelman HE (2000) Mononuclear phagocytes mediate blood-brain barrier compromise and neuronal injury during HIV-1-associated dementia. J Leukoc Biol 68:413–422
Pulliam L (2014) Cognitive consequences of a sustained monocyte type 1 IFN response in HIV-1 infection. Curr HIV Res 12:77–84
Pulliam L, Gascon R, Stubblebine M, McGuire D, McGrath MS (1997) Unique monocyte subset in patients with AIDS dementia. Lancet 349:692–695
Pulliam L, Calosing C, Sun B, Grunfeld C, Rempel H (2014) Monocyte activation from interferon-α in HIV infection increases acetylated LDL uptake and ROS production. J Interferon Cytokine Res 34:822–828
Roberts ES, Burudi EME, Flynn C, Madden LJ, Roinick KL, Watry DD, Zandonatti MA, Taffe MA, Fox HS (2004) Acute SIV infection of the brain leads to upregulation of IL6 and interferon-regulated genes: expression patterns throughout disease progression and impact on neuroAIDS. J Neuroimmunol 157:81–92
Saini M, Potash MJ (2014) Chronic, highly productive HIV infection in monocytes during supportive culture. Curr HIV Res 12:317–324
Sandler NG, Douek DC (2012) Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol 10:655–666
Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, Pedersen C, Ruxrungtham K, Lewin SR, Emery S, Neaton JD, Brenchley JM, Deeks SG, Sereti I, Douek DC, Group ISS (2011) Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis 203:780–790
Sas AR, Bimonte-Nelson H, Smothers CT, Woodward J, Tyor WR (2009) Interferon-alpha causes neuronal dysfunction in encephalitis. J Neurosci 29:3948–3955
Shan L, Siliciano RF (2013) From reactivation of latent HIV-1 to elimination of the latent reservoir: the presence of multiple barriers to viral eradication. Bioessays 35:544–552
Shiramizu B, Ananworanich J, Chalermchai T, Siangphoe U, Troelstrup D, Shikuma C, De Grutolla V, Sithinamsuwan P, Praihirunkit P, Rattanamanee S, Valcour V, Group SS (2012) Failure to clear intra-monocyte HIV infection linked to persistent neuropsychological testing impairment after first-line combined antiretroviral therapy. J Neurovirol 18:69–73
Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF (2003) Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9:727–728
Smurzynski M, Wu K, Letendre S, Robertson K, Bosch RJ, Clifford DB, Evans S, Collier AC, Taylor M, Ellis R (2011) Effects of central nervous system antiretroviral penetration on cognitive functioning in the ALLRT cohort. AIDS 25:357–365
Stevenson M (2014). Role of myeloid cells in HIV-1-host interplay. J Neurovirol
Tan J, Sattentau QJ (2013) The HIV-1-containing macrophage compartment: a perfect cellular niche? Trends Microbiol 21:405–412
Tavazzi E, Morrison D, Sullivan P, Morgello S, Fischer T (2014) Brain inflammation is a common feature of HIV-infected patients without HIV encephalitis or productive brain infection. Curr HIV Res 12:97–110
Thompson KA, Cherry CL, Bell JE, McLean CA (2011) Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol 179:1623–9
Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, Suwanwela NC, Jagodzinski L, Michael N, Spudich S, van Griensven F, de Souza M, Kim J, Ananworanich J (2012) Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis 206:275–282
Valcour VG, Ananworanich J, Agsalda M, Sailasuta N, Chalermchai T, Schuetz A, Shikuma C, Liang C-Y, Jirajariyavej S, Sithinamsuwan P, Tipsuk S, Clifford DB, Paul R, Fletcher JLK, Marovich MA, Slike BM, DeGruttola V, Shiramizu B, Team SP (2013) HIV DNA reservoir increases risk for cognitive disorders in cART-naïve patients. PLoS One 8:e70164
Vojnov L, Martins MA, Bean AT, Veloso de Santana MG, Sacha JB, Wilson NA, Bonaldo MC, Galler R, Stevenson M, Watkins DI (2012) The majority of freshly sorted simian immunodeficiency virus (SIV)-specific CD8(+) T cells cannot suppress viral replication in SIV-infected macrophages. J Virol 86:4682–4687
Walker JA, Sulciner ML, Nowicki KD, Miller AD, Burdo TH, Williams KC (2014) Elevated numbers of CD163+ macrophages in hearts of simian immunodeficiency virus-infected monkeys correlate with cardiac pathology and fibrosis. AIDS Res Hum Retrovir 30:685–694
Watters SA, Mlcochova P, Gupta RK (2013) Macrophages: the neglected barrier to eradication. Curr Opin Infect Dis 26:561–566
Wei DG, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, Hesselgesser J, Irrinki A, Murry JP, Stepan G, Stray KM, Tsai A, Yu H, Spindler J, Kearney M, Spina CA, McMahon D, Lalezari J, Sloan D, Mellors J, Geleziunas R, Cihlar T (2014) Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog 10:e1004071
Williams K, Schmitz J, Kuroda M, Alvarez X, Klein R, Simmone M, al. e (2001a). CD8 T lymphocytes control the development of SIV encephalitis: implications for the neuropathogenesis of AIDS. Abstract from International Society of Neuroimmunology Sixth International Congress, Edinburgh, Scotland Sept 3–7, 2001. J Neuroimmunol 118: 384
Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA (2001b) Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med 193:905–915
Wong JK, Hezareh M, Günthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD (1997) Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291–1295
Yearley JH, Pearson C, Carville A, Shannon RP, Mansfield KG (2006) SIV-associated myocarditis: viral and cellular correlates of inflammation severity. AIDS Res Hum Retrovir 22:529–540
Zalar A, Figueroa MI, Ruibal-Ares B, Baré P, Cahn P, de Bracco MM, Belmonte L (2010) Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Anticancer Res 87:269–271
Ziegler-Heitbrock L, Lotzerich M, Schaefer A, Werner T, Frankenberger M, Benkhart E (2003) IFN-alpha induces the human IL-10 gene by recruiting both IFN regulatory factor 1 and Stat3. J Immunol 171:285–290
Financial conflicts of interest
The authors declare that they have no financial conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is supported by grants from the NIMH and NIDA, National Institutes of Health, to JR and DJV.
Rights and permissions
About this article

Cite this article
Rappaport, J., Volsky, D.J. Role of the macrophage in HIV-associated neurocognitive disorders and other comorbidities in patients on effective antiretroviral treatment. J. Neurovirol. 21, 235–241 (2015). https://doi.org/10.1007/s13365-015-0346-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-015-0346-y