
Abstract
Herpes simplex encephalitis (HSE) remains the most important cause of fatal sporadic encephalitis in man. Caused by herpes simplex virus type 1 (HSV-1), and more rarely by HSV-2, it can have devastating clinical consequences for the patient, especially when the instigation of acyclovir therapy has been delayed by more than 2 days or more. Even with acyclovir treatment, nearly a third of patients may die or suffer significant morbidity. Both host and viral factors may interact to affect the clinical phenotype. Here we consider some of the recently published management guidelines for HSE and comment on various current issues of contention. The latter includes the timing and frequency of cerebrospinal fluid examinations for the polymerase chain reaction detection of HSV, decisions regarding acyclovir therapy including the consequences of delay in its initiation, and the use of corticosteroids in the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction and neuropathogenesis
Herpes simplex encephalitis (HSE) is an important acute neurological infection not only because is it the commonest fatal sporadic encephalitis occurring in man but also since it is treatable with the antiviral drug acyclovir, and treatment delays are associated with a significantly poorer disease outcome (Solomon et al. 2012). Here we consider some current issues in this area while not attempting to provide a comprehensive overview. The disease is caused by the herpes simplex virus (HSV), a pathogenic human herpesvirus, the genome of which can be detected in 85–90 % of trigeminal ganglia in individuals at autopsy (Baringer 2000), an observation that is consistent with a very high seroprevalence of HSV-1 of around 90 % in normal asymptomatic individuals (Kennedy and Chaudhuri 2002). It has always been difficult to reconcile this extensive viral latency carriage with the relative rarity of HSE itself, and this is a longstanding conundrum in this field. Thus, while precise incidence figures are difficult to obtain, it has been estimated that HSE accounts for almost 20 % of all cases of encephalitis (Granerod et al. 2010) and has an annual incidence of 1 in 250,000 to 500,000 (Solomon et al. 2012). HSV-1 causes 90 % of adult cases of HSE with 10 % caused by HSV-2; the latter is also a commoner cause in neonates and the immunosuppressed (Chaudhuri and Kennedy 2002; Miller et al. 2013).
HSE typically affects the frontal and temporal lobes, accounting for the characteristic clinical features including personality changes, cognitive impairment, aphasia, seizures, and focal weakness (Kennedy and Chaudhuri 2002; Whitley and Gnann 2002), but in rare cases, the brainstem may be preferentially involved (Livorsi et al. 2010). The disease can sometimes affect both hemispheres simultaneously (Sureka and Jakkani 2012). It seems likely that the virus is localized to these specific brain regions as a consequence of spread along particular neural pathways, possibly via the olfactory pathways to the temporal lobes, or from the trigeminal ganglia, where the virus is latent, to the frontal and temporal lobes (Davis and Johnson 1979; Baringer 2000). The etiology of HSE is not known at present though we have several clues as to disease neuropathogenesis, and there has been some recent investigation into this question. While it seems logical to assume that host factors such as age and the level of immunocompetence are key determinants of disease, the question on variability of herpesvirus neurovirulence as a causative factor also has to be taken into consideration, and possibly host and viral factors may interact to cause a particular disease phenotype. However, based on a small number of cases, a seminal study in 1982 compared the brain and oral–labial HSV-1 isolates in patients with HSE and concluded, on the basis of restriction enzyme analysis of these, that HSE may occur as a consequence not only of an expected viral reactivation but also from a primary or a secondary HSV-1 infection (Whitley et al. 1982). Recently, the entire genome sequence was obtained in both an HSV-1 isolate from an encephalitic patient and a laboratory strain (Szpara et al. 2010). Sequence differences were detected between these two strains with a number of potentially causal genes determining variations in clinical phenotype. These considerations might favor the concept that HSE is due to primary infection rather than reactivation (Steiner 2011). This approach demonstrates the potential for deep genome sequencing to provide the potential molecular basis for particular HSV-1 (and other viruses) to produce neurological disease, and more extensive studies of this kind are awaited (Szpara et al. 2010).
It has been shown that about 20 % of HSE cases, diagnosed by cerebrospinal fluid (CSF) polymerase chain reaction (PCR) (see below) can present as a relatively mild or atypical disease (Fodor et al. 1998). Though there were quite a small number reported, these mild cases were associated with either HSV-1 or HSV-2 and frequently associated with immunosuppression. How frequently HSE may present as mild disease in the general population remains to be determined, but host factors are clearly critical. Recently, specific genetic, rather than general, host factors have been observed to be associated with the development of HSE in children (Lafaille et al. 2012; Herman et al. 2012). Thus, it was shown that HSE in two children was the consequence of an autosomal recessive deficiency in the intracellular protein UNC-93B leading to impaired cellular interferon (IFN) responses (Casrouge et al. 2006). Subsequent work has confirmed that HSE in children may result from single gene errors in Toll-like receptor 3 (TLR3)-IFN type 1 and 3 pathways (Pérez de Diego et al. 2013). Why such genetic determinants should manifest in childhood but not in adult HSE is unclear.
Guidelines for management of encephalitis
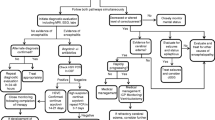
There has been sizeable interest in recent years in producing general guidelines for the investigation and management of viral encephalitis with an understandable emphasis on HSE. There is considerable merit in such documents, which should, by definition, be evidence-based wherever possible, particularly when there is a general variation in clinical practice, some issues are unclear, and where prompt and appropriate treatment is vital but not always implemented. A simple algorithm for management of suspected HSE was suggested by one of us in 2004 (Kennedy 2004), and the authors produced (with others) a comprehensive, strongly evidence-based guideline document of the European Federation of Neurological Societies (EFNS) for diagnosis and management of viral meningoencephalitis in 2005 (Steiner et al. 2005) which was then updated in 2010 (Steiner et al. 2010). Many of the suggestions were already in common practice, and the extent to which these were actually implemented in Europe is unknown. Partly because of the lack of guideline implementation, but also because of the great importance of the subject to both general physicians and neurologists, Solomon and colleagues recently produced a clear and comprehensive national guideline document of the Association of British Neurologists (ABN) and British Infection Association on the management of suspected viral encephalitis (Solomon et al. 2012). This document, which is evidence-based and includes a management algorithm similar in part to, but more complex and detailed than, our previous one, addresses directly the key management issues that clinicians face and is clear and unequivocal in its many recommendations. We will briefly address here some of the recommendations, many of which are, inevitably and understandably, based on expert opinion rather than hard evidence gleaned from randomized clinical trials (Thwaites 2012).
There is a general issue about the potential legal implications of clinical guidelines. If recommendations are evidence-based, then clearly there is an obligation for the clinician to follow them. However, if they are not, and are based primarily on expert opinion, then while it is certainly wise to follow them as far as possible, the approach should always be determined by the particular clinical situation.
Considerations in the investigation of suspected HSE
The most immediate clinical challenge is to diagnose HSE when it exists, as early treatment is available and vital, and there is a very close relationship between diagnostic and treatment considerations in HSE. Examination of the CSF should always be carried out if at all possible. While the CSF typically shows a lymphocytosis of 10–200/mm3 (or more) and a raised protein of 0.5–1.0 g/l or more (Kennedy and Chaudhuri 2002; Solomon et al. 2012), the mainstay of diagnosis is detection of HSV by PCR. The evidence base for the utility of CSF PCR in HSE and other viral encephalitides has been recently reviewed in detail in a separate EFNS-ENS guideline document (Steiner et al. 2012). The sensitivity of CSF PCR for detecting HSV DNA is about 96 %, and the specificity in experienced laboratories is about 99 % (Steiner et al. 2012). Neuroimaging is also of great diagnostic importance in suspected HSE. While it is preferred to carry out neuroimaging, ideally with an MRI scan, as soon as possible in cases of suspected viral encephalitis, this should certainly not delay starting acyclovir. The MRI is abnormal in almost all cases of HSE showing the characteristic abnormalities of edema and/or abnormal enhancement in one or both temporal and frontal lobes, the insular cortex, and the angular gyrus (Baringer 2000; Steiner et al. 2010). The other key importance of neuroimaging (and a CT scan may be carried out if MRI is not available) is to exclude a space-occupying lesion such as a tumor or abscess or evidence of significant cerebral edema that would make a lumbar puncture potentially hazardous. The diagnosis of HSE may be complicated by a number of important timing issues. If there is to be a delay of more than 6 h before starting acyclovir, and provided that there is no clinical contraindication to performing a lumbar puncture, the recent ABN guidelines recommend examining the CSF even if neuroimaging has not been performed. A pitfall, based on a few reports, is that the CSF PCR may be negative for HSV-1 during the first 3 days of the illness (Steiner et al. 2012). If so, and if the clinical suspicion of HSE continues, then the CSF should be reexamined after a few days as the PCR may then become positive. The chance of a positive PCR is highest during the first week, even when the patient is on acyclovir, following which the viral yield falls (Lakeman and Whitley 1995). Consequently, the CSF PCR may also be negative in definite HSE if it is examined too late in the illness, after 10–14 days. Clinicians need to be very aware of these timing issues when assessing the patient with suspected HSE. Another contentious issue is whether to repeat the CSF after 14 days of acyclovir as is recommended by the ABN guidelines and, which is carried out in some, but certainly not all, neurological units. We do not routinely repeat the PCR test on CSF at 14 days, unless there is a clinical indication that the patient is not recovering as quickly as would be expected, and there is still a strong clinical suspicion of ongoing HSE, and especially if the first CSF sample had been taken within 3 days and was negative. Though repeating the CSF PCR routinely after 14 days of treatment was also recommended in a prior European consensus report (Cinque 1996), at present we lack an evidence base for carrying this out routinely.
Considerations in the HSE patient treated with acyclovir
As soon as the diagnosis of HSE is suspected, the patient should be commenced on intravenous (IV) acyclovir of 10 mg/kg three times daily for 14 days. Caution should be exercised if there is known renal impairment. In the immunocompromised patient, or in children under 12 years, acyclovir should be continued for at least 21 days (Solomon et al. 2012). Untreated, the mortality from HSE is about 70 %, whereas following acyclovir treatment, it is reduced to about 28 % at 18 months (Whitley and Gnann 2002), but morbidity still remains high. While acyclovir therapy was clearly an enormous advance, nevertheless these complications are still far from ideal. One important issue is when it is safe to stop acyclovir when the CSF PCR has been negative. Our view is that it is safe to do so when an alternative diagnosis has been unambiguously established, making acyclovir unnecessary. However, the recent ABN guidelines extend this approach by including two other criteria: (a) if the CSF PCR is negative on two occasions 24–48 h apart and MRI does not suggest HSE and (b) if the CSF PCR is negative once, more than 72 h after onset of symptoms, with normal consciousness level, a normal MRI, and a CSF white cell count <5/mm3 (Solomon et al. 2012). These are expert opinions, and we would exercise caution with this second criterion in the absence of an alternative diagnosis, and for criterion a, would want to ensure that the second CSF specimen has been taken more than 3 days after the onset of symptoms.
Though in our experience most clinicians are generally quick to instigate acyclovir therapy for suspected HSE, it is still the case that in some instances treatment is unfortunately delayed, potentially resulting in severe sequelae. Is it possible to define a cutoff period in such delay for the development of serious complications? Evidence suggests that a delay of 48 h or more in starting acyclovir results in a worse prognosis. Thus, in one study on 42 HSE patients, it was found that the time from admission to the start of acyclovir treatment was longer in patients with a poor outcome, with 1.8 days in the good outcome group and 4.0 days for the poor outcome group (McGrath et al. 1997). In another study on 85 HSE patients, it was found that a delay of >2 days between admission to hospital and initiation of acyclovir therapy was found to be independently associated with a poor outcome (Raschilas et al. 2002). Possible reasons for a delay in starting acyclovir were found in another retrospective study on 184 HSE patients, which include severe underlying disease, alcohol abuse, and a delay of >1 day from admission to the first brain imaging (Poissy et al. 2009). An interesting question is whether it is possible to correlate the degree of disability with the number of days for which acyclovir treatment has been delayed. While the 48 h of acyclovir delay cutoff point has a clear evidence base, we do not think that it is possible at present to say that one can accurately predict the degree of ensuing disability from the number of days of delay, especially when comparing, say, 3 days with 4 days. However, we know that HSE patients below 30 years with a Glasgow coma score of >6 who are treated with acyclovir within 4 days of the onset of symptoms have a good outcome (Whitley and Gnann 2002), so there is very good reason to believe that a delay in treatment of >4 days will be deleterious.
Another issue of contention is the routine use of corticosteroids in HSE. At present, there is no existing evidence on giving steroids to all patients with HSE, and we do not advocate this, a view that was also expressed in the ABN guidelines (Solomon et al. 2012). In theory, steroids could dampen down the neuroinflammatory response in HSE and be beneficial. By contrast, administering steroids could potentially exacerbate a CNS viral infection, making their use hazardous. The one situation where steroid administration is certainly appropriate is when an HSE patient shows cerebral edema that could both contraindicate lumbar puncture and result in severe brain swelling, coning, and death. In that emergency situation, urgent neurosurgical decompression may be lifesaving. At present, some clinicians do give steroids to their HSE patients, while most probably do not. While one retrospective study on 45 patients indicated that HSE patients who were not treated with steroids had a worse outcome than those who had received them (Kamei et al. 2005), a large prospective randomised trial on the use of steroids in HSE will be required to answer this question definitively. Notwithstanding these caveats, it is probably unlikely that an immunocompetent patient with HSE given steroids for the first few days along with acyclovir would suffer from steroid-associated complications.
An interesting question is whether a course of oral valacyclovir following the 14-day IV administration of acyclovir should be given to improve the final outcome after infection. At present, again, we just do not know the answer to this question, but the ongoing NIAID Collaborative Antiviral Study Group trial should be able to provide an answer to this question in due course. It should be added that we do not believe that it is justified to give orally rather than IV acyclovir during the first 14-day course. Not only is there no evidence base for this, but also we are unable to conceive how it could be ethically justified to test this in HSE patients. Since acyclovir has only a 15–39 % oral absorption, valaciclovir, an acyclovir prodrug for oral administration with a better bioavailability, was introduced (Field and Vere Hodge 2013). Penciclovir achieves higher intracellular concentrations than acyclovir, and famciclovir is a prodrug of penciclovir with better oral bioavailability. None, however, has replaced acyclovir, so far, for the treatment of HSE.
The question of acyclovir resistance in the immunocompetent patient has been raised by many observers in this area. This has been described (Schulte et al. 2010), but we believe that it must be a very rare event at present, despite the widespread use of acyclovir, including for many patients who turn out not to have HSE. In a single case report (Schulte et al. 2010), a 27-year-old immunocompetent woman with HSE was shown to have been infected with an HSV-1 resistant to acyclovir due to a mutation in the thymidine kinase gene. Treatment with the second-line antiviral drug foscarnet led to a clinical recovery.
Conclusions
The pathogenesis of HSE is still elusive, and the mechanism responsible for HSV penetration into the brain parenchyma, unknown. Irrespective of this, the disorder requires immediate diagnosis and therapy in order to reduce or prevent permanent neurological damage. PCR analysis of CSF is a major tool in establishing the diagnosis, but when a CSF sample is not available or when the PCR is negative for HSV in the context of a suggestive clinical context, acyclovir therapy should be commenced as soon as possible while the effort to look for an alternative diagnosis continued (Table 1).
References
Baringer JR (2000) Herpes simplex virus encephalitis. In: Davis LE, Kennedy PGE (eds) Infectious diseases of the nervous system. Butterworth-Heinemann, Oxford, pp 139–164
Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, Alcais A, Picard C, Mahfoufi N, Nicolas N, Lorenzo L, Plancoulaine S, Sénéchal B, Geissmann F, Tabeta K, Hoebe K, Du X, Miller RL, Héron B, Mignot C, de Villemeur TB, Lebon P, Dulac O, Rozenberg F, Beutler B, Tardieu M, Abel L, Casanova JL (2006) Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314:308–312
Chaudhuri A, Kennedy PGE (2002) Diagnosis and treatment of viral encephalitis. Postgrad Med J 78:575–583
Cinque P, Cleator GM, Weber T, Monteyne P, Sindic CJ, van Loon AM (1996) The role of laboratory investigation in the diagnosis and management of patients with suspected herpes simplex encephalitis: a consensus report. The EU concerted action on virus meningitis and encephalitis. J Neurol Neurosurg Psychiatry 61:339–345
Davis LE, Johnson RT (1979) An explanation for the localization of herpes simplex encephalitis? Ann Neurol 5:2–5
Field HJ, Vere Hodge RA (2013) Recent developments in anti-herpesvirus drugs. Br Med Bull 106(1):213–249
Fodor PA, Levin MJ, Weinberg A, Sandberg E, Sylman J, Tyler KL (1998) Atypical herpes simplex virus encephalitis diagnosed by PCR amplification of viral DNA from CSF. Neurology 51:554–559
Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D, Cunningham R, Zuckerman M, Mutton KJ, Solomon T, Ward KN, Lunn MP, Irani SR, Vincent A, Brown DW, Crowcroft NS, UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group (2010) Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 10:835–844
Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Pérez de Diego R, Abhyankar A, Israelsson E, Guo Y, Cardon A, Rozenberg F, Lebon P, Tardieu M, Heropolitanska-Pliszka E, Chaussabel D, White MA, Abel L, Zhang SY, Casanova JL (2012) Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 209:1567–1582
Kamei S, Sekizawa T, Shiota H, Mizutani T, Itoyama Y, Takasu T, Morishima T, Hirayanagi K (2005) Evaluation of combination therapy using aciclovir and corticosteroid in adult patients with herpes simplex virus encephalitis. J Neurol Neurosurg Psychiatry 76:1544–1549
Kennedy PGE (2004) Viral encephalitis: causes, differential diagnosis, and management. J Neurol Neurosurg Psychiatry 75(Suppl 1):i10–i15
Kennedy PGE, Chaudhuri (2002) Herpes simplex encephalitis. J Neurol Neurosurg Psychiatry 73:237–238
Lakeman FD, Whitley RJ (1995) Diagnosis of herpes simplex encephalitis: application of polymerase chain reaction to cerebrospinal fluid from brain-biopsied patients and correlation with disease. National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. J Infect Dis 171:857–863
Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, Ordovas-Montanes J, Jouanguy E, Plancoulaine S, Tu E, Elkabetz Y, Al-Muhsen S, Tardieu M, Schlaeger TM, Daley GQ, Abel L, Casanova JL, Studer L, Notarangelo LD (2012) Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 491:769–773
Livorsi D, Anderson E, Qureshi S, Howard M, Wang YF, Franco-Paredes C (2010) Brainstem encephalitis: an unusual presentation of herpes simplex virus infection. J Neurol 257:1432–1437
McGrath N, Anderson NE, Croxson MC, Powell KF (1997) Herpes simplex encephalitis treated with acyclovir: diagnosis and long term outcome. J Neurol Neurosurg Psychiatry 63:321–326
Miller S, Mateen FJ, Aksamit AJ Jr (2013) Herpes simplex virus 2 meningitis: a retrospective cohort study. J Neurovirol 19:166–171
Pérez de Diego R, Mulvey C, Crawford M, Trotter MW, Lorenzo L, Sancho-Shimizu V, Abel L, Zhang SY, Casanova JL, Godovac-Zimmermann J (2013) The proteome of Toll-like receptor 3-stimulated human immortalized fibroblasts: implications for susceptibility to herpes simplex virus encephalitis. J Allergy Clin Immunol 131:1157–1166
Poissy J, Wolff M, Dewilde A, Rozenberg F, Raschilas F, Blas M, Georges H, Chaffaut C, Yazdanpanah Y (2009) Factors associated with delay to acyclovir administration in 184 patients with herpes simplex virus encephalitis. Clin Microbiol Infect 15:560–564
Raschilas F, Wolff M, Delatour F, Chaffaut C, De Broucker T, Chevret S, Lebon P, Canton P, Rozenberg F (2002) Outcome of and prognostic factors for herpes simplex encephalitis in adult patients: results of a multicenter study. Clin Infect Dis 35:254–260
Schulte EC, Sauerbrei A, Hoffmann D, Zimmer C, Hemmer B, Mühlau M (2010) Acyclovir resistance in herpes simplex encephalitis. Ann Neurol 67:830–833
Solomon T, Michael BD, Smith PE, Sanderson F, Davies NW, Hart IJ, Holland M, Easton A, Buckley C, Kneen R, Beeching NJ, National Encephalitis Guidelines Development and Stakeholder Groups (2012) Management of suspected viral encephalitis in adults–Association of British Neurologists and British Infection Association National Guidelines. J Infect 64:347–373
Steiner I (2011) Herpes simplex virus encephalitis: new infection or reactivation? Curr Opin Neurol 24:268–274
Steiner I, Budka H, Chaudhuri A, Koskiniemi M, Sainio K, Salonen O, Kennedy PG (2005) Viral encephalitis: a review of diagnostic methods and guidelines for management. Eur J Neurol 12:331–343
Steiner I, Budka H, Chaudhuri A, Koskiniemi M, Sainio K, Salonen O, Kennedy PG (2010) Viral meningoencephalitis: a review of diagnostic methods and guidelines for management. Eur J Neurol 17:999–e57
Steiner I, Schmutzhard E, Sellner J, Chaudhuri A, Kennedy PG (2012) EFNS-ENS guidelines for the use of PCR technology for the diagnosis of infections of the nervous system. Eur J Neurol 19:1278–1291
Sureka J, Jakkani RK (2012) Clinico-radiological spectrum of bilateral temporal lobe hyperintensity: a retrospective review. Br J Radiol 85:e782–e792
Szpara ML, Parsons L, Enquist LW (2010) Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J Virol 84:5303–5313
Thwaites GE (2012) The management of suspected encephalitis. BMJ 344:e3489
Whitley RJ, Gnann JW (2002) Viral encephalitis: familiar infections and emerging pathogens. Lancet 359:507–513
Whitley R, Lakeman AD, Nahmias A, Roizman B (1982) DNA restriction-enzyme analysis of herpes simplex virus isolates obtained from patients with encephalitis. N Engl J Med 307:1060–1062
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kennedy, P.G.E., Steiner, I. Recent issues in herpes simplex encephalitis. J. Neurovirol. 19, 346–350 (2013). https://doi.org/10.1007/s13365-013-0178-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-013-0178-6