
Abstract
The fundamental question posed here is why in dorsal root ganglia herpes simplex viruses (HSV) can establish a silent infection in which only latency associate transcripts (LAT) and miRNAs are expressed and the neuronal cell survives whereas in non-neuronal cells HSV replicates and destroys the infected cells. Current evidence indicates that in productive infection there are two checkpoints. The first is at activation of α genes and requires a viral protein (VP16) that recruits HCF-1, Oct1, LSD1, and the CLOCK histone acetyl transferase to demethylate histones and initiate transcription. The second checkpoint involves activation of β and γ genes. An α protein, ICP0, activates transcription by displacing HDAC1 or 2 from the HDAC/CoREST/LSD1/REST repressor complex at its DNA binding sites. Current data suggest that in dorsal root ganglia VP16 and HCF-1 are not translocated to neuronal nucleus and that the HDAC/CoREST/LSD1/REST complex is not suppressed—a first step in silencing of the viral genome and establishment of heterochromatin. The viral genome remains in a state of equilibrium with respect to viral gene expression. The function of both LAT and the micro RNAs is to silence low level expression of viral genes that could reactivate the latent genomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The fundamental question posed by virologists for more than seven decades is the mechanism by which herpes simplex viruses (HSV) periodically flare up and cause recrudescent lesions. In the intervening decades, we have learned that at the portal of entry into the body, HSV-1 causes a productive infection in which infected cells are destroyed and the virus multiplies and a latent infection in dorsal root and autonomic ganglia in which the virus is silenced and the neuron survives (Roizman et al. 2007). Thus, the original question doubled: How does a virus that is prone to infect and destroy cells at the portal of entry become silent in neurons and what makes it reactivate?
The two posed questions are not operationally defined and in fact are based on a false premise. The underlying assumption is that HSV finds resistance to replication in the neurons but not at the portal of entry, or to simplify the conceptualization of the problem, that it is sufficient for the viral DNA to present itself in the nucleus for viral replication to ensue in cells at the portal of entry. The hypothesis tested in this laboratory is based on an entirely different premise, namely that the virus encounters resistance to infection in cells at the portal of entry into the body and in the neurons, that it is able to overcome resistance at the portal of entry into the body but not in neurons (Roizman 2011). The corollary of this hypothesis is that, by a stochastic process, the silencing in some neurons ceases or is overcome and the virus replicates. Finally, following the principle of Occam’s razor, the tests of this hypothesis should focus at least initially on the assumption that the same cellular factors that operate at the portal of entry of the virus into the body also operate in neurons in which the viral DNA is silenced. The objective of this report is to provide a brief summary of the tests of this hypothesis.
The checkpoints of viral replication in productively infected cells
The fundamental hypothesis underlying these studies and stated above predicts that cells attempt to silence viral DNA on entry into the nucleus. Several lines of evidence indicate that this is indeed the case. On entry into the cells, HSV DNA is coated by histones and other proteins (Knipe and Cliffe 2008; Kutluay and Triezenberg 2009; Lacasse and Schang 2010). This finding per se does not prove resistance to infection if only a fraction of viral DNA actually initiates replication. The non-replicating DNA most likely ends as heterochromatin. The evidence that the bound proteins actually attempt to repress viral DNA comes from two series of experiments. Thus, nearly 30 years ago, it was shown that a viral protein, VP16 or α-TIF, activates transcription of α genes—the first set of genes expressed after infection (reviewed in Roizman et al. 2007; Roizman 2011). The current version of the events illustrated in Fig. 1 is that the complex VP16, Oct1, and the host transcriptional factor HCF1 binds to response elements in promoters of α genes and recruits LSD1 (lysine-specific demethylase) and the CLOCK histone acetyl transferase to remodel chromatin in order to initiate transcription (Liang et al. 2009; Zhou et al. 2010; Kalamvoki and Roizman 2010, 2011; Metzger et al. 2005). It is noteworthy that LSD1 is unstable in the absence of its partner, CoREST (Shi et al. 2005. Yang et al. 2006; Lee et al. 2005). CoREST in turn interacts with REST. In non-neuronal cells, CoREST/REST proteins form a complex with HDAC1 or HDAC2 and act as repressors of neuronal genes (reviewed in Ballas and Mandel 2005; Tapia-Ramírez et al. 1997; Gopalakrishnan 2009). The key recent evidence on the role of these proteins in initiation of transcription of viral genes rests on studies showing that even a partial depletion of LSD1, CoREST, REST, or CLOCK by corresponding siRNA resulted in a delay in the initiation of transcription of α genes (Zhou et al. 2010). The fundamental conclusion is that the host attempts to repress the viral genome from the moment of entry of the virus into cells and that a viral tegument protein, VP16, carried by the virion into the cell recruits the cellular factors essential to initiate transcription of α genes

A model of the events at the two key checkpoints in viral gene expression during productive infection. a Schematic representation of infected cell at the point of entry of viral DNA and VP16 into the nucleus. b–e Schematic representations of nuclei of infected cells. b HSV DNA is bound by histones and repressors including the HDAC-1/CoREST/REST complex. c Assembly of transcriptional machinery at response elements in promoter of α genes. VP16 recruits Oct1, HCF-1, and LSD1. Included in the complex are CLOCK histone acetyl transferase and additional transcriptional factors that enable the expression of α genes. d Newly made α protein ICP0 binds to CoREST and dislodges HDAC1 or 2 enabling the expression of β and γ genes
The second set of experiments also has a long history. It has been known for many years that an α protein designated as the infected cell protein no. 0 (ICP0) is a promiscuous transactivator of genes introduced into cells by transfection or infection. At low multiplicities of infection, α genes are expressed but the transition from α to β (early) or γ genes did not ensue (reviewed in Roizman et al. 2007; Roizman 2011). The block in the transition from α to β and γ gene expression could be overcome at high multiplies of infection. ICP0 differed from ICP4, another regulatory α protein required for expression of viral genes (Roizman et al. 2007). Unlike ICP4, ICP0 does not bind to viral DNA. A clue to the role of ICP0 in the transition from α to β gene expression emerged from the observation that approximately 70 residues of ICP0 located close to the C terminus of the protein were also present at the N terminus of CoREST (Gu et al. 2005; Gu and Roizman 2007, 2009). Subsequent studies showed that CoREST and ICP0 interact that the binding site for CoREST in ICP0 is next to the sequence homologous to the N terminus of CoREST. In CoREST, the binding site for ICP0 is at the N terminus and corresponds to the sequence that is conserved in ICP0 (Gu and Roizman 2007). In addition, two findings solidified a functional role of the interaction between ICP0 and the HDAC1 or 2/CoREST/REST repressor complex. Thus, ICP0 dislodged HDAC1-1 from the CoREST/REST complex. In addition, at a slightly later time in infection, at least a fraction of the dislodged HDAC1, CoREST, and REST were exported to the cytoplasm (Gu et al. 2005). The key experiment designed to determine the role of the interaction was based on the expectation that if the role of ICP0 is to block repression by the HDAC-1 or 2/CoREST/REST complex, then a dominant negative CoREST incapable of binding HDAC1 should compensate for the absence of ICP0 in cells infected with a mutant virus at low multiplicities of infection (Gu and Roizman 2009). This was in fact the case: A mutant virus in which ICP0 was replaced by a dominant negative CoREST replicated to 10- to 100-fold higher titers than ΔICP0 mutant in a cell-type-dependent fashion.
The fundamental conclusions of the two series of studies are that the viral genome is repressed immediately upon entry into the nucleus that expression of all viral genes requires derepression of the genome in two stages: the first one for the expression of α genes and the second for the expression of β and γ genes. It is noteworthy that the same repressor components are involved at both checkpoints. An obvious question is why has the virus evolved a two-step process of derepression of the viral genome when in fact a single step would have sufficed?
Derepression of the HSV genome to enable post-α genes expression is but one of the functions of ICP0. This multifunctional protein localizes early in infection at ND10 nuclear bodies. Here it acts as an E3 ubiquitin ligase to degrade promyelocytic leukemia protein and SP100 (Boutell et al. 2002; Hagglund et al. 2002; Gu and Roizman 2003). A key objective accomplished by degrading ICP0 is to render the infected cell resistant to exogenous interferon (Chee et al. 2003). ICP0 also binds to Bmal1 and helps recruit Bmal1 and its partner, CLOCK, to the ND10 bodies (Kawaguchi et al. 2001; Kalamvoki and Roizman 2010). The various functions of ICP0 appear to be interdependent (Gu and Roizman 2009).
The latent state
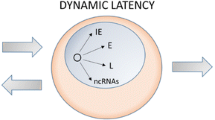
There is universal agreement that in experimental animal systems, HSV infects nerve endings at the site of inoculation. The sequelae are less clear. The evidence indicates that the virus replicates at least in some cells that replication ultimately abates and the residual virus detected in ganglia is in latently infected neurons (Roizman et al. 2007). It is convenient to explore the hypothesis illustrated schematically in Fig. 2 that the virus replicates in some neurons that are ultimately destroyed but is silenced in other neurons. A key finding that bears on the silencing of viral DNA in neurons committed to the maintenance of latent state is the observation that VP16 and HCF1 are both retained in the neuronal cytoplasm and do not reach the nucleus (Boissiere et al. 1999; Kristie et al. 1999). Presumably in the absence of VP16 and HCF1 α gene would not be expressed. A central issue is whether the absence of HCF1 and VP16 alone accounts for the silencing of viral DNA in latently infected neurons.

A model of the establishment of latency in neurons. a Viral DNA enters nucleus of the neuron in the absence of VP 16 or HCF-1 or both. b Viral DNA is silenced by histones and repressors. c Viral DNA–host protein complex is remodeled into heterochromatin. LAT and miRNAs carryout surveillance to degrade accumulating low level transcripts of key viral genes
Parsimony requires that we consider that the response of neuronal cells in dorsal root ganglia to the entry of HSV DNA into the nuclei is the same as that of cells at the portal of entry. According to this hypothesis in latently infected neurons, HSV fails to initiate transcription at two rather than solely at one checkpoint. Thus, the lack of VP16 and HCF1 would preclude the breaching of the first checkpoint. The absence of α proteins and in particular ICP0 would preclude the expression of β and γ genes. The test of this hypothesis rests on the demonstration that the second checkpoint exists in neurons. The data in support of the hypothesis are a mutant virus consisting of the wild-type genome into which a dominant negative REST genes driven by the SV40 promoter multiplied to higher levels and was more lethal than the wild-type parent virus (Du et al. 2010). In mice infected with this mutant by the corneal route, the trigeminal ganglia were totally destroyed. The results of the experiment suggest that a larger fraction of neurons initially committed to silencing of DNA are induced to replicate the virus through inactivation of checkpoint 2 by the dominant negative REST. The conclusion to be drawn from this study is: HSV does not invariably overcome checkpoints 1 or 2 consistent with the prediction that unchecked replication with increased morbidity and mortality is detrimental to virus spread in nature.
The second hypothesis that silencing of viral DNA in latently infected neurons is due to synthesis of LATs and associated miRNAs is fundamentally untenable as stated. The LAT is a stable intron derived from a very much longer short-lived precursor RNA (Wu et al. 1996). Most of the miRNAs identified to date derive from the domain of LAT or the larger RNA from which it is derived (Cui et al. 2006; Tang et al. 2008; Umbach et al. 2008, 2009). Notwithstanding the loss of neurons associated with infections with ΔLAT viruses, these mutants do establish latent infections albeit not as efficiently as wild-type viruses (Javier et al. 1988; Wang et al. 2001.) A more appropriate role of LATs and miRNAs is surveillance and degradation of low level transcription of viral genes from the silenced DNA. The failure of such surveillance and rapid degradation would result in a low level spontaneous reactivation and destruction of neurons.
It has been reported that in latently infected neurons, HSV DNA is in heterochromatin and that the fraction of remodeled DNA correlates with the expression of LATs (Chen et al. 2002; Maillet et al. 2006; Neumann et al. 2007; Kwiatkowski et al. 2009; Wang et al. 2005). Conceptually, sequestration of HSV DNA in heterochromatin could silence viral gene expression. There are two fundamental issues: Foremost, viral DNA would be expected to interact with appropriate repressors and is silenced in the course of its remodeling into heterochromatin. The key questions are what precipitates and are the key steps in the remodeling of viral DNA. Second, a vexing problem that threatens to remain unresolved for some time is that only a fraction of latent HSV DNA reactivates. Is the state of heterochromicity uniform for all HSV DNA?
As noted in the beginning of this section, the unfolded scenario of events taking place in infected ganglia makes the assumption that both productive and latent infection occur simultaneously and independently of each other. Direct evidence in support of this conclusion is scant. The conclusion is based on the observation that in cell culture expression of α genes invariably leads to cell death even under conditions in which viral replication does not ensue. We cannot exclude the hypothesis that the initial events in infected neurons are similar if not identical and that a stochastic event forces a lytic rather than silent infection.
Reactivation: principles and problems
In humans, clinical reactivations more appropriately called recrudescences of herpetic vesicles follow certain stimuli such as fever, exposure to UV light, and emotional or hormonal stimulation. It is now abundantly clear that reactivations of virus replication are far more frequent than the recrudescence of herpetic vesicles (Roizman and Sears 1987). The prevailing thought is that the immune system precludes the reactivated virus from causing recrudescent lesions. The fundamental question is the mechanism of reactivation. There are two interrelated issues.
The first issue stems from the abundant evidence that to initiate gene expression, VP16/OCT1/HCF1 must bind to promoters of α genes and induce the synthesis of α protein. VP16 is unlikely to be present to initiate replication of latent virus in neurons that have been harboring viral DNA for many decades. Hence, either (a) a host factor substitutes for VP16 to initiate α gene expression, (b) in neurons VP16 is expressed first followed by activation of α genes as suggested by Thompson et al. 2009 or (c) the promoter of the gene encoding ICP0 contains elements that enable it to be expressed at a low but sufficient level in the absence of VP16. Current evidence does not fully discriminate between the possibilities enumerated above.
The second issue is what triggers reactivation regardless of the pathway by which it takes place. An intriguing possibility is that in neurons HCF1 resides in the cytoplasm but is transported to the nucleus following stress (Kristie et al. 1999). This hypothesis would support a role for a newly synthesized VP16, but it would also imply that transcription of the VP16 genes and entry of HCF1 are functionally linked.
Conclusions
Deconstruction of the HSV latent state presents interesting challenges not insuperable by current methodology. The central issue is whether the models used to dissect the latent state embraces all elements of the human model.
References
Ballas N, Mandel G (2005) The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol 15:500–506
Boissiere S, Hughes T, O’Hare P (1999) HCF-dependent nuclear import of VP16. EMBO J 18:480–489
Boutell C, Sadis S, Everett RD (2002) Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol 76:841–850
Chee AV, Lopez P, Pandolf PP, Roizman B (2003) Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J Virol 77:7101–7105
Chen S, Lee L, Garber D, Shaffer P, Knipe D, Coen D (2002) Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J Virol 76:4764–4772
Cui C, Griffiths A, Li G, Silva LM, Kramer MF, Gaasterland T, Wang XJ, Coen DM (2006) Prediction and identification of herpes simplex virus 1-encoded microRNAs. J Virol 80:5499–5508
Du T, Zhou G, Khan S, Gu H, Roizman B (2010) Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc Natl Acad Sci USA 2010(107):15904–15909
Gopalakrishnan V (2009) REST and the RESTless: in stem cells and beyond. Futur Neurol 4:317–329
Gu H, Roizman B (2003) The degradation of PML and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc Natl Acad Sci USA 100:8963–8968
Gu H, Roizman B (2007) Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST/REST complex. Proc Natl Acad Sci USA 104:17134–17139
Gu H, Roizman B (2009) The two functions of HSV-1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J Virol 83:181–187
Gu H, Liang Y, Mandel G, Roizman B (2005) Components of the REST/CoREST/histone deacetylase repressor are disrupted, modified and translocated in HSV-1-infected cells. Proc Natl Acad Sci USA 102:7571–7576
Hagglund R, Van Sant C, Lopez P, Roizman B (2002) Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc Natl Acad Sci USA 99:631–666
Javier RT, Stevens JG, Dessette VB, Wagner EK (1988) A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology 166:254–257
Kalamvoki M, Roizman B (2010) The circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus genes expression. Proc Natl Acad Sci USA 107:17721–17726
Kalamvoki M, Roizman B (2011) The histone acetyl transferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27 and ICP22. J Virol 85:9472–9477
Kawaguchi Y, Tanaka M, Yokoymama A, Matsuda G, Kato K, Kagawa H, Hirai K, Roizman B (2001) Herpes simplex virus 1 alpha regulatory protein ICP0 functionally interacts with cellular transcription factor BMAL1. Proc Natl Acad Sci USA 98:1877–1882
Knipe DM, Cliffe AR (2008) Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221
Kristie TM, Vogel JL, Sears AE (1999) Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc Natl Acad Sci USA 96:1229–1233
Kutluay SB, Triezenberg SJ (2009) Regulation of histone deposition on the herpes simplex virus type 1 genome during lytic infection. J Virol 83:5835–5845
Kwiatkowski DL, Thompson HW, Bloom DC (2009) The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181
Lacasse JJ, Schang LM (2010) During lytic infections, herpes simplex virus type 1 is in complexes with properties of unstable nucleosomes. J Virol 84:1920–1933
Lee MG, Wynder C, Cooch N, Shiekhattar R (2005) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437:432–435
Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM (2009) Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med 15:1312–1317
Maillet S, Naas T, Crepin S, Roque-Afonso AM, Lafay F, Efstathiou S, Labetoulle M (2006) Herpes simplex virus type 1 latently infected neurons differentially express latency-associated and ICP0 transcripts. J Virol 80:9310–9321
Metzger E, Wissmann M, Yin N, Muller J, Schneider R, Peters AHFM, Gunther T, Buettner S, Schule R (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437:436–439
Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM (2007) In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol 81:13248–13253
Roizman B (2011) The checkpoints of viral gene expression in productive and latent infection: the role of the HDAC/CoREST/LSD1/REST repressor complex. J Virol 85:7474–7482
Roizman B, Sears AE (1987) An inquiry into the mechanism of herpes simplex virus latency. Annu Rev Microbiol 41:543–571
Roizman B, Knipe DM, Whitley RJ et al (2007) Herpes simplex viruses. In: Knipe DM (ed) Fields virology, 5th edn. Lippincott Williams & Wilkins, New York, pp 2501–2601
Shi YJ, Matson C, Iwase S, Baba T, Shi Y (2005) Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19:857–864
Tang S, Bertke AS, Patel A, Wang K, Cohen JI, Krause PR (2008) An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc Natl Acad Sci USA 105:10931–10936
Tapia-Ramírez J, Eggen BJ, Peral-Rubio MJ, Toledo-Aral JJ, Mandel G (1997) A single zinc finger motif in the silencing factor REST represses the neural-specific type II sodium channel promoter. Proc Natl Acad Sci USA 94:1177–1182
Thompson RL, Preston CM, Sawtell NM (2009) De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog 5:e1000352
Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR (2008) MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783
Umbach JL, Nagel M, Cohrs R, Gilden D, Cullen BR (2009) Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J Virol 83:10677–10683
Wang K, Pesnicka L, Guancial E, Krause P, Straus S (2001) The 2.2-kilobase latency-associated transcript of herpes simplex virus type 2 does not modulate viral replication, reactivation, or establishment of latency in transgenic mice. J Virol 75:8166–8172
Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM (2005) Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci USA 102:16055–16059
Wu T, Su Y, Block T, Taylor J (1996) Evidence that two latency-associated transcripts of herpes simplex virus type 1 are nonlinear. J Virol 70:5962–5967
Yang M, Gocke C, Luo X, Borek D, Tomchick D, Machius M, Otwinowski Z, Yu H (2006) Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell 23:377–387
Zhou G, Du T, Roizman B (2010) The CoREST/REST repressor is both necessary and inimical for expression of herpes simplex virus gene expression. MBio 2:e00313-10
Acknowledgments
These studies were aided by a grant from the National Cancer Institute grant R37 CA78766.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Roizman, B., Zhou, G. & Du, T. Checkpoints in productive and latent infections with herpes simplex virus 1: conceptualization of the issues. J. Neurovirol. 17, 512–517 (2011). https://doi.org/10.1007/s13365-011-0058-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-011-0058-x