
Abstract
Fourier transform ion cyclotron resonance (FTICR) mass spectrometry provides unparalleled mass measurement accuracy and resolving power. However, propagation of the technique into new analytical fields requires continued advances in instrument speed and sensitivity. Here, we describe a substantial redesign of our custom-built 9.4 tesla FTICR mass spectrometer that improves sensitivity, acquisition speed, and provides an optimized platform for future instrumentation development. The instrument was designed around custom vacuum chambers for improved ion optical alignment, minimized distance from the external ion trap to magnetic field center, and high conductance for effective differential pumping. The length of the transfer optics is 30% shorter than the prior system, for reduced time-of-flight mass discrimination and increased ion transmission and trapping efficiency at the ICR cell. The ICR cell, electrical vacuum feedthroughs, and cabling have been improved to reduce the detection circuit capacitance (and improve detection sensitivity) 2-fold. The design simplifies access to the ICR cell, and the modular vacuum flange accommodates new ICR cell technology, including linearized excitation, high surface area detection, and tunable electrostatic trapping potential.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Fourier transform ion cyclotron resonance mass spectrometry (FTICR MS) offers the highest resolving power m/Δm 50% (Δm 50% is the full peak width at half-maximum peak height) and mass measurement accuracy of any mass analyzer [1, 2]. Unparalleled peak capacity allows resolution and identification of thousands of components in a complex mixture in a single mass spectrum with sub-part per million (ppm) mass measurement accuracy [3]. For bottom-up and top-down proteomics [4, 5], high mass measurement accuracy increases identification confidence and narrows database search space, and ultrahigh resolving power allows separation of overlapping isotopic distributions and enables charge state determination of large, multiply charged biomolecules [6–8]. FTICR is uniquely suited to analysis of complex natural organic mixtures, such as petroleum crude oil [9] and humic/fulvic acids [10]. Ultrahigh mass resolving power (m/Δm 50% ≥ 400,000) is needed to resolve thousands of peaks in a petroleum mass spectrum [11], and ultrahigh mass measurement accuracy is needed for confident elemental composition assignments [12]. However, the number of unresolved multiplets increases at higher m/z due to decreased resolving power and increased variation in possible elemental compositions. The complexity of current applications thus requires further optimization and advancement of FTICR MS technology.
In FTICR-MS, ions are typically accumulated external to the magnetic field at a pressure many orders of magnitude greater than that required for image charge analysis in the ICR cell [13]. The physical dimensions of the magnet and the need for multiple stages of differential pumping necessitate an ion flight path of approximately one meter between the ion accumulation trap and the ICR cell, leading to time-of-flight mass discrimination and decreased trapping efficiency during ion transfer to the ICR cell [14]. The trapped ions are excited to a large cyclotron orbit by resonant RF voltage and subsequently observed by image charge detection. Longer time-domain transient lifetime is needed for greater resolving power, to resolve isobaric species and to assign elemental composition at high m/z [15, 16]. Coherent cyclotron motion of an ion packet is required to obtain high-resolution mass spectra. However, the number of coherently orbiting ions can diminish with time due to electric and magnetic field inhomogeneities and ion cloud interactions [17, 18].
The continuing development of higher magnetic field instruments increases several figures of merit, such as data acquisition speed, resolving power, reduced sensitivity to space charge effects, mass measurement accuracy, and dynamic range [6, 19]. However, many other instrument parameters besides higher magnetic field strength can be modified to improve instrument performance [20], e.g., increased ion transfer and trapping efficiency, and new ICR cell geometries that more closely approach harmonicity in the electrostatic trapping potential at large cyclotron radius [21–23]. Increased ion cyclotron radius can reduce space charge frequency shifts [24], and precise control of the number of trapped ions can reduce variation in space charge frequency shifts from successive ion injections [25]. In addition, the signal magnitude is directly proportional to the proximity of the ions to the detection electrodes, so the closer the ion cloud is to the ICR cell, the greater the signal induced on the detection electrodes and the greater the sensitivity. However, deviation from ideal magnetic and electric field, ion-neutral collisions, and ion-image charge interactions increase with post-excitation cyclotron radius and limit transient lifetime and induce additional frequency shifts [26–29]. The optimal post-excitation cyclotron radius depends on the required resolving power and therefore will vary with application [30].
Here we describe a substantial redesign and initial performance of a 9.4 tesla FTICR mass spectrometer. The prior instrument utilized the same 9.4 tesla magnet and generated numerous publications over the past 12 years [13, 31], setting several benchmarks for FTICR technology [32–34]. Prior modifications include the addition of a quadrupole mass filter and electron capture dissociation (ECD) capability, while the back end of the instrument (ion transfer optics, vacuum system, ICR cell) remained unchanged. The present modifications are designed to shorten the ion flight path from the external accumulation ion trap to ICR cell, improve optical alignment along the center of the bore of the magnet, increase accessibility to the ICR cell for future modifications, decrease detection circuitry capacitance for improved signal-to-noise and detection sensitivity, and facilitate complex experiments and future modifications.
2 Experimental
2.1 Sample Preparation
Standard compounds (melittin, Ultramark, MRFA peptide, caffeine, and bovine apo-transferrin) were purchased from Sigma Aldrich (St. Louis, MO, USA) and used without further purification. Samples were dissolved in HPLC grade water at 1 mg/mL. Electrosprayed samples were diluted to 1–2 μM in 49:49:2 methanol/water/acetic acid or 49.75:49.75:0.5 methanol/water/formic acid. Direct infusion was performed at a flow rate of 0.5 μL/min. Middle Eastern crude oil was diluted to 0.25 mg/mL in toluene and infused at 50 μL/min into a modified ThermoFisher source [3] for atmospheric pressure photoionization.
2.2 Magnetic Field
The instrument is based on a passively shielded 9.4 tesla superconducting horizontal solenoid magnet (Oxford Instruments, Oxney Mead, UK) with a 225 mm room temperature bore diameter. The measured magnetic field inhomogeneity is 5 ppm peak-to-peak variation on the surface of a cylinder 40 mm in diameter and 80 mm along the symmetry (z-) axis of the magnetic field. Repeated measurements with and without the ICR cell and electrical feedthroughs installed confirmed that the chosen materials have no measurable impact on the magnetic field.
2.3 Vacuum System
Figure 1 shows a schematic diagram of the instrument. The vacuum system has been designed around custom vacuum chambers (Nor-Cal Products, Yreka, CA, USA), thereby shortening the distance from the exit of the ion accumulation region to the magnetic field center by 30% from the prior design (distance in new instrument=1.4 m). Concentricity and parallelism of the critical conflat flanges of the new chambers enable superior optical alignment along the central axis of the instrument relative to the prior design. Specifically, alignment of the magnet, vacuum chambers, and ion optics with a diode laser resulted in estimated residual misalignment of less than 300 μm from the ion accumulation octopole (octopole 2) to the center of the ICR cell flange. The aluminum vacuum chamber (Atlas Technologies, Port Townsend, WA, USA) extends through the bore of the magnet and incorporates explosively-bonded bimetal (aluminum and titanium) conflat flanges for robust UHV operation with standard copper gaskets. Custom high conductance elbows (Johnsen Ultravac, Burlington, Ontario, Canada) efficiently couple three high-speed turbomolecular pumps (1,100 L/s) to the UHV chambers. The custom elbows allow optimal placement of the pumps with respect to the fringe magnetic field as well as a shorter ion flight path to the ICR cell.

Schematic diagram of the NHMFL 9.4 tesla hybrid quadrupole FTICR mass spectrometer. Conductance-limiting apertures are labeled “CL”.
2.4 Ion Transfer
Ions created at atmospheric pressure are transferred through different pressure regions by rf-only octopoles (1.6 mm diameter, 4.8 mm i.d. titanium rods) operated at 1.5 to 2.0 MHz and 190 < V p-p < 240 rf amplitude. Ions pass through a quadrupole mass filter and accumulate in octopole 2 before transfer to the ICR cell. Helium gas is typically leaked into the accumulation region to maintain a pressure of 1 mTorr, but other gases (e.g., nitrogen or argon) are easily substituted. Appropriate acceleration of ions into octopole 2 can achieve collisionally activated dissociation (CAD). To produce more efficient fragmentation for proteins and peptides, the collision gas is switched to nitrogen. Octopole 2 has been modified with angled wire electrodes for improved ion extraction efficiency [35]. A mini-UHV gate valve with 2 ¾ in. Conflat edges isolates the source region from the UHV region of the instrument. The distance between octopole 3 and conductance limit 4 (CL4) is set at 7 mm so that the UHV gate valve can operate freely. Based on signal magnitude, a negligible loss (<10%) in ion transmission occurs as ions traverse that gap compared to the prior 1 mm separation between octopole 3 and CL4. A single long transfer octopole has been divided into two individual transfer octopoles biased at −60 V and operated at 2.4 MHz and 250 V p-p with a custom-built rf tuned circuit (Ardara Technologies, North Huntingdon, PA, USA). The two transfer octopoles are wired separately so that individual power supplies may be used for each rod set. The reduced capacitance of the separate octopoles compared to a single long octopole increases the available range of rf frequency and V p-p amplitude.
2.5 ICR Cell
There are three stages of differential pumping in the UHV region of the instrument. The first two are located on the source side of the magnet, and the final stage is located on the back side of the magnet. The base pressure in the ion detection region is <2 × 10–10 Torr as measured by a Bayard-Alpert ionization gauge. The ICR cell (constructed from oxygen-free high conductivity (OFHC) copper with no additional treatment to the internal surfaces) is supported through the flange on the back UHV chamber, allowing for convenient access for modification of the ICR cell while maintaining optical alignment of the vacuum system along the center of the magnetic field. The ICR cell geometry is open cylindrical, with three equal sections of 100 mm in length and an inner diameter of 94 mm [36]. The angular extents of the detection and excitation electrodes have been modified to be 120° and 60° [37]. The gaps between all adjacent cell electrodes are 2.5 mm. The cell is wired externally for capacitive coupling [38]. Ions are frequency-sweep excited (from high to low frequency) to ~40% of the cell radius. The post-excitation radius is optimized to minimize ion-ion interactions while maintaining ion cloud phase coherence for >5 seconds to achieve the resolving power required for analysis of complex mixtures. The waveform for dipolar excitation is input from the arbitrary waveform generator to a high power rf amplifier (ENI, Rochester, NY, USA) followed by a center tapped transformer (Northhills, Syosett, NY, USA) to split the waveform by 180° and provide additional rf voltage gain. Available ion fragmentation methods in the ICR cell include electron capture dissociation (ECD) [39] and infrared multi-photon dissociation (IRMPD) [40]. A heated dispenser cathode is located on the magnetic field axis. A 40 W continuous IR laser (Synrad, Mukilteo, WA, USA) is mounted off-axis at an angle of 4° relative to the central axis of the system. The laser beam intersects the central axis of the system at approximately the center of the ICR cell.
2.6 Data Generation and Handling
Instrument control, data acquisition, and data analysis are carried out with a modular ICR data station [41]. The time-domain ICR signal is Hanning apodized, zero-filled once, and Fourier transformed to produce a magnitude-mode spectrum that is converted to a mass-to-charge ratio by a two term calibration equation [42].
3 Results and Discussion
3.1 Ion Transfer
Ions are initially accumulated in octopole 2 and are collected in the ICR cell by gated trapping for subsequent excitation and detection. The decreased length of the vacuum system shortens the ion flight time, which is defined as the time period between ion ejection from octopole 2 (induced by a voltage pulse applied to the extraction wires) and voltage increase of the ICR cell front trapping electrodes. Figure 2 shows the signal magnitude at different flight times for various m/z species following application of 30 V to the angled extraction wires of octopole 2 for prior and present instrument configurations. The optimal flight times are shorter in the new system and there is greater overlap in residence time between the high and low m/z species in the ICR cell. The new system is thus less sensitive to time-of-flight mass discrimination. In addition, the distribution of flight times for ions of the same m/z is narrowed. The shorter flight distance decreases the time period during which ions of the same m/z but different axial kinetic energy can separate throughout the transfer event, resulting in increased trapping efficiency. The most direct method to reduce TOF effects is to reduce the ion flight length between the ion accumulation trap and the ICR cell. Other reported methods include use of multiple transfer events with different ion flight periods [14, 43, 44], segmentation of the transfer multipole to provide axial containment throughout the ion transfer event [45], conversion of axial into radial energy as the ions enter the ICR cell (sidekick) [46], or m/z-dependent ion velocity adjustment during the transfer event [47, 48].

FTICR signal magnitude as a function of flight time (see text) for ions of m/z ranging from 195 to 1622. (a) The previous instrument and (b) the new instrument, with 30 V applied to the extraction wires in both cases. (c) Further decrease in m/z-dependent time-of-flight discrimination is achieved by lowering the extraction wire voltage to 15 V.
The user-defined flight time determines which ions are trapped and which are lost. With low voltage (5–15 V) applied to the extraction electrodes, ions initially at different axial position and velocity in the wired octopole are ejected at sufficiently different times to induce broadening of the arrival time distribution for each m/z, and it is more likely that heavy m/z ions that are immediately ejected from the accumulation trap reach the ICR cell at the same time as light ions that were ejected at a later time. However, a broader arrival time distribution reduces the trapping efficiency at the ICR cell for that species. Increased voltage on the extraction electrodes (~30 V) shortens the duration of ion ejection from the accumulation cell so that all ions are ejected at approximately the same time, thereby narrowing the arrival time distribution and the m/z range trapped in the ICR cell for a given flight time. Thus, the detected m/z range for ions will be determined by the acceleration voltage and the length of time between ion extraction from octopole 2 and the closing of the front trapping electrodes of the ICR cell. Lower voltage on the wires is preferred for detection of ions spanning a broad m/z range, whereas higher voltage is preferred for ions of narrow m/z range or for focusing of ions of a particular m/z, as for selective injection of precursor ions for in-cell fragmentation (ECD or IRMPD). Figure 2c illustrates the wider m/z distribution that is trapped when a lower voltage (15 V rather than 30 V) is applied to the extraction electrodes.
3.2 Detection Circuitry
The number of ions needed to create a detectable signal is directly related to the capacitance of the detection circuitry, including the ICR cell plates, electrical feedthroughs, and cabling [49]. Reduction in capacitance of the system results in an increase in detected ICR signal magnitude. The new ICR cell has been designed with larger gaps between all cell plates to decrease capacitance. The wire that carries the image current from the detection plates to the back flange is shielded for noise reduction and the inner diameter of that grounded shield has been increased to 11 mm to minimize capacitance. The total capacitance of the detection circuitry is thus reduced by 2-fold from 145 pF to 70 pF, thereby doubling the ICR signal strength. Ideally, capacitance could be further reduced by placement of the preamplifier in vacuum adjacent to the ICR cell. However, outgassing, magnetic susceptibility, heat dissipation, and ease of operation limit implementation of that option.
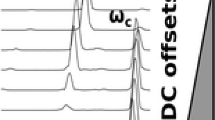
Figure 3 shows the increase signal-to-noise ratio with decreasing detection capacitance. The circuit capacitance was varied by introducing different lengths of BNC cabling between the back flange and the preamplifier. Figure 3 (inset) shows the signal-to-noise ratio at two different capacitance settings. The signal amplitude exhibits the theoretically predicted 3-fold increase in signal magnitude corresponding to 3-fold decrease in capacitance. The noise level remained unaffected by the lower capacitance. The inner diameter of the ICR cell is 94 mm, approximately 56% larger than for commercial ICR cells (~60 mm i.d.). The larger diameter cell was chosen to exploit the large bore and homogenous magnetic field of the magnet. In addition, the larger ICR cell has a greater ion charge capacity: ions can spread out further axially and radially (for ions excited to the same fraction of the cell radius) for reduced space charge frequency shifts.

Signal-to-noise ratio as a function of detection circuit capacitance, C. The solid black line represents a 1/C fit, consistent with theory. The inset shows the signal magnitude for detection circuit capacitance of 87 pf (red) or 266 pf (black).
3.3 Peptide and Protein Analysis
Accurate mass is essential for unambiguous identification of biological molecules, and their fragmentation can provide complete structural characterization and site(s) of post-translational modification(s) [50]. Ion fragmentation techniques available for the present instrument include CAD in the accumulation octopole, and ECD and IRMPD inside the ICR cell. Figure 4 illustrates IRMPD and ECD for the melittin (M+4 H)4+ precursor ion. The precursor ions were isolated by the quadrupole mass filter and accumulated externally before transfer to the ICR cell for ECD or IRMPD. The improved ion transfer efficiency greatly reduces the accumulation period required to yield a sufficient number of precursor ions in the ICR cell. Note that even with careful mechanical alignment of the ion optics and ICR cell with the magnetic field axis, ECD efficiency still depends strongly on correct phasing of electron injection with respect to ion magnetron motion [51].

MS/MS of melittin. The 4+ charge state is isolated by use of the mass-selective quadrupole (inset) and accumulated. Ions are transferred to the ICR cell and fragmented by either IRMPD (top) or ECD (bottom).
Mass analysis of intact proteins is inherently more difficult due to distribution of the analyte signal over a greater number of charge states, wider isotopic distribution, and adducts (cations, buffers, etc.), all of which decrease the signal magnitude of all measured species. Higher charge states also result in increased ion–ion Coulombic repulsion. MS/MS of these proteins results in a larger number of overlapping isotopic distributions, requiring high resolution to directly determine the charge states and masses of the fragment ions. Accurate determination of the masses of many fragment ions is needed to confidently assign sequence and post-translational modifications. In top-down analysis, large proteins are typically fraction-collected and analyzed off-line for target analysis, so that it is possible to signal-average a large number of acquisitions to generate useful fragment ion information [52]. However, high resolution, high mass measurement accuracy MS and MS/MS data acquired on-line for intact proteins of 5–40 kDa have recently been reported [53, 54].
Figure 5 shows top-down analysis of a large protein. A quadrupole isolation window (m/z range≈25, Figure 5, top) selectively accumulates the 54+ charge state from direct infusion of non-reduced bovine transferrin (~78,000 Da). The inset demonstrates baseline resolution of isotopomers differing by ~1 Da), without the need to iteratively tune instrument parameters such as ion cyclotron excitation amplitude, ion cooling period, or trapping potential. Collisional dissociation is achieved by ion acceleration into the accumulation octopole in the presence of nitrogen gas, (Figure 5, bottom). The voltage applied to the extraction wires in the accumulation octopole was increased to 35 V and the ion transfer period was adjusted to achieve optimal transfer efficiency. The most abundant products provide sequence tags of similar length and two different charge states, from which the protein may readily be identified.

FTICR mass spectra of bovine transferrin. Top: The inset shows baseline resolution of the 54+ charge state with >180,000 resolving power at m/z 1445 (quadrupole isolation, 3 s external ion accumulation, 30 averaged spectra). Bottom: Top-down analysis by collisional fragmentation in the accumulation octopole, providing similar sequence tags for two different charge states.
3.4 Complex Mixture Analysis
The analysis of compositional complex samples such as petroleum [9] or natural organic matter [10] requires high resolving power and mass measurement accuracy with a large dynamic range over a wide mass range. Ultrahigh resolution is needed to resolve the thousands of peaks in a single mass spectrum, resulting in >60 peaks per nominal mass [3], and to resolve mass doublets such as 12C3 vs. 32SH4 whose masses differ by only 3.4 mDa. Such resolution requires that the time-domain ICR signal persist essentially undamped for at least 3 s. Moreover, the fixed charge capacity of the ICR cell is distributed over thousands of m/z values, thereby severely limiting dynamic range for a single data acquisition. Thus, multiple (>100) time-domain acquisitions are typically averaged to obtain a single spectrum with acceptable signal-to-noise ratio and a dynamic range of 103–104. For complex mixtures, the new instrument achieves comparable signal-to-noise ratio at 8-fold lower sample concentration and with half the ion external accumulation time period.
Figure 6 illustrates a typical positive-ion atmospheric pressure photoionization (APPI) FTICR mass spectrum of a whole crude oil. The m/z distribution is wider than that typically observed on the prior instrument due to shorter ion flight path, so that the spectrum extends to lower m/z values for better representation of a molecular weight distribution that matches that of an ion trap mass spectrum (LTQ mass spectrometer, ThermoFisher Corp., Bremen, Germany). Mass spectral peak magnitude is ideally proportional to analyte concentration. Therefore, the molecular weight distribution for a complex mixture is typically verified by comparison to that from a different mass spectrometer that is not subject to TOF mass discrimination. The improved transfer efficiency at lower m/z extends the mass range for assigned elemental compositions. The inset shows some mass splits commonly observed for petroleum. The resolution is Fourier-limited: i.e., the transient persists essentially undamped for the entire data acquisition period (5.6 s), extending the upper mass limit at which 12C3 vs. 32SH4 doublets are resolved. For APPI, even the 1.1 mDa mass doublet (SH 133 C1 vs. C4) is baseline-resolved (m/Δm50%≈800,000). Elemental compositions in petroleum naturally form homologous series, providing for identification of components up to ~1,000 Da [55].

Positive-ion atmospheric pressure photoionization FTICR MS of a Middle Eastern light crude oil, acquired with the new (top panel) and old (middle panel) instrument. The inset illustrates resolution of commonly observed mass splits of 1.1 and 3.4 mDa. An ion trap mass spectrum (bottom panel) provides a molecular weight distribution without TOF mass discrimination.
4 Conclusions
The design and initial performance of our custom-built 9.4 tesla FTICR mass spectrometer are described. Improvements over the prior design increase sensitivity and overall performance of the instrument. The custom vacuum chambers reduce the ion transfer distance by 30% for decreased time-of-flight mass discrimination and improved ion transfer and trapping efficiency at the ICR cell. A reduction in the capacitance of the detection circuitry results in a 2-fold increase in sensitivity. The instrument is ideally suited for proteomic analysis of large intact proteins with the capability to isotopically resolve large intact proteins, and features fragmentation by IRMPD and ECD in the ICR cell as well as CAD in the external accumulation octopole. The instrument readily achieves the resolving power, high dynamic range, and mass measurement accuracy needed for complex mixture analysis. For the same signal-to-noise ratio, petroleum samples may now be analyzed at 1/8 the concentration and 1/2 the ion external accumulation period relative to the prior instrument. The current instrument modular configuration facilitates implementation of new technology. Future efforts will focus on further reduction in detection circuit capacitance for improved sensitivity, and exploration of new ICR cell configurations that approach ideal 3D axial quadrupolar trapping potential.
References
Comisarow, M.B., Marshall, A.G.: Fourier Transform Ion Cyclotron Resonance Spectroscopy. Chem. Phys. Lett. 25, 282–283 (1974)
Marshall, A.G., Hendrickson, C.L., Jackson, G.S.: Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: A Primer. Mass Spectrom. Rev. 17, 1–35 (1998)
Purcell, J.M., Hendrickson, C.L., Rodgers, R.P., Marshall, A.G.: Atmospheric Pressure Photoionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry for Complex Mixture Analysis. Anal. Chem. 78, 5906–5912 (2006)
Bogdanov, B., Smith, R.D.: Proteomics by FTICR Mass Spectrometry: Top Down and Bottom Up. Mass Spectrom. Rev. 24, 168–200 (2005)
Mann, M., Kelleher, N.L.: Precision Proteomics: The Case for High Resolution and High Mass Accuracy. Proc. Natl. Acad. Sci. U.S.A. 105, 18132–18138 (2008)
Schaub, T.M., Hendrickson, C.L., Horning, S., Quinn, J.P., Senko, M.W., Marshall, A.G.: High-Performance Mass Spectrometry: Fourier Transform Ion Cyclotron Resonance at 14.5 Tesla. Anal. Chem. 80, 3985–3990 (2008)
Hicks, L., Weinreb, P., Konz, D., Marahiel, M.A., Walsh, C.T., Kelleher, N.L.: Fourier-Transform Mass Spectrometry for Detection of Thioester-Bound Intermediates in Unfractionated Proteolytic Mixtures of 80 and 191 Kda Portions of Bacitracin a Synthetase. Anal. Chim. Acta 496, 217–224 (2003)
Zubarev, R.A., Hakansson, P., Sundqvist, B.: Accuracy Requirements for Peptide Characterization by Monoisotopic Molecular Mass Measurements. Anal. Chem. 68, 4060–4063 (1996)
Marshall, A.G., Rodgers, R.P.: Petroleomics: Chemistry of the Underworld. Proc. Natl. Acad. Sci. U.S.A. 105, 18090–18095 (2008)
Stenson, A.C., Marshall, A.G., Cooper, W.T.: Exact Masses and Chemical Formulas of Individual Suwannee River Fulvic Acids from Ultrahigh Resolution Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectra. Anal. Chem. 75, 1275–1284 (2003)
Panda, S.K., Andersson, J.T., Schrader, W.: Mass-Spectrometric Analysis of Complex Volatile and Nonvolatile Crude Oil Components: A Challenge. Anal. Bioanal. Chem. 389, 1329–1339 (2007)
Kim, S., Rodgers, R.P., Marshall, A.G.: Truly "Exact" Mass: Elemental Composition can be Determined Uniquely from Molecular Mass Measurement at.Apprx.0.1mDa Accuracy for Molecules up to Approximately 500 Da. Int. J. Mass Spectrom. 251, 260–265 (2006)
Senko, M.W., Hendrickson, C.L., Emmett, M.R., Shi, S.D.H., Marshall, A.G.: External Accumulation of Ions for Enhanced Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J. Am. Soc. Mass Spectrom. 8, 970–976 (1997)
Wong, R.L., Amster, I.J.: Combining Low and High Mass Ion Accumulation for Enhancing Shotgun Proteome Analysis by Accurate Mass Measurement. J. Am. Soc. Mass Spectrom 17, 205–212 (2006)
Kind, T., Fiehn, O.: Metabolomic Database Annotations Via Query of Elemental Compositions: Mass Accuracy is Insufficient Even at Less Than 1 ppm. BMC Bioinformatics 7, 234 (2006)
Marshall, A.G., Rodgers, R.P.: Petroleomics: The Next Grand Challenge for Chemical Analysis. Acc. Chem. Res. 37, 53–59 (2004)
Peurrung, A.J., Kouzes, R.T.: Long-Term Coherence of the Cyclotron Mode in a Trapped Ion Cloud. Phys. Rev. E Stat. Phys. (Plasmas, Fluids, and Related Interdisciplinary Topics) 49, 4362–4368 (1994)
Gordon, E.F., Muddiman, D.C.: Impact of Ion Cloud Densities on the Measurement of Relative Ion Abundances in Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: Experimental Observations of Coulombically Induced Cyclotron Radius Perturbations and Ion Cloud Dephasing Rates. J.f Mass Spectrom. 36, 195–203 (2001)
Marshall, A.G., Guan, S.: Advantages of High Magnetic Field for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Rapid Commun. Mass Spectrom. 10, 1819–1823 (1996)
Kaiser, N.K., Skulason, G.E., Weisbrod, C.R., Bruce, J.E.: A Novel Fourier Transform Ion Cyclotron Resonance Mass Spectrometer with Improved Ion Trapping and Detection Capabilities. J. Am. Soc. Mass Spectrom. 20, 755–762 (2009)
Weisbrod, C.R., Kaiser, N.K., Skulason, G.E., Bruce, J.E.: Trapping Ring Electrode Cell: A FTICR Mass Spectrometer Cell for Improved Signal-to-Noise and Resolving Power. Anal. Chem. 80, 6545–6553 (2008)
Tolmachev, A.V., Robinson, E.W., Wu, S., Kang, H., Lourette, N.M., Pasa-Tolic, L., Smith, R.D.: Trapped-Ion Cell with Improved DC Potential Harmonicity for FT-ICR MS. J. Am. Soc. Mass Spectrom. 19, 586–597 (2008)
Boldin, K.A., Nikolaev, E.N.: Fourier Transform Ion Cyclotron Resonance Cell with Dynamic Harmonization of the Electric Field in the Whole Volume by Shaping of the Excitation and Detection Electrode Assembly. Rapid Commun. Mass Spectrom. 25, 1–5 (2011)
Easterling, M.L., Mize, T.H., Amster, I.J.: Routine Part-Per-Million Mass Accuracy for High-Mass Ions: Space-Charge Effects in MALDI FT-ICR. Anal. Chem. 71, 624–632 (1999)
Syka, J.E.P., Marto, J.A., Bai, D.L., Horning, S., Senko, M.W., Schwartz, J.C., Ueberheide, B., Garcia, B., Busby, S., Muratore, T., Shabanowitz, J., Hunt, D.F.: Novel Linear Quadrupole Ion Trap/FT Mass Spectrometer: Performance Characterization and Use in the Comparative Analysis of Histone H3 Post-Translational Modifications. J. Proteome Res. 3, 621–626 (2004)
Chen, S.P., Comisarow, M.B.: Simple Physical Models for Coulomb-Induced Frequency Shifts and Coulomb-Induced Inhomogeneous Broadening for Like and Unlike Ions in Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Rapid Commun. Mass Spectrom. 5, 450–455 (1991)
Leach Franklin, E.I.I.I., Kharchenko, A., Heeren Ron, M.A., Nikolaev, E., Amster, I.J.: Comparison of Particle-In-Cell Simulations with Experimentally Observed Frequency Shifts Between Ions of the Same Mass-To-Charge in Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J. Am. Soc. Mass Spectrom. 21, 203–208 (2010)
Gorshkov, M.V., Marshall, A.G.: Analysis and Elimination of Systematic Errors Originating from Coulomb Mutual Interaction and Image Charge in Fourier Transform Ion Cyclotron Resonance Precise Mass Difference Measurements. J. Am. Soc. Mass Spectrom. 4, 855–868 (1993)
Arkin, C.R., Laude, D.A.: Collision Induced Ion Ejection in an FTICR Trapped-Ion Cell. J. Am. Soc. Mass Spectrom. 16, 422–430 (2005)
Tolmachev, A.V., Robinson, E.W., Wu, S., Pasa-Tolic, L., Smith, R.D.: FT-ICR MS Optimization for the Analysis of Intact Proteins. Int. J. Mass Spectrom. 287, 32–38 (2009)
Senko, M.W., Hendrickson, C.L., Pasa-Tolic, L., Marto, J.A., White, F.M., Guan, S., Marshall, A.G.: Electrospray Ionization Fourier Transform Ion Cyclotron Resonance at 9.4 T. Rapid Commun. Mass Spectrom. 10, 1824–1828 (1996)
Shi, S.D.H., Hendrickson, C.L., Marshall, A.G.: Counting Individual Sulfur Atoms in a Protein by Ultrahigh-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: Experimental Resolution of Isotopic Fine Structure in Proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 11532–11537 (1998)
He, F., Hendrickson, C.L., Marshall, A.G.: Baseline Mass Resolution of Peptide Isobars: A Record for Molecular Mass Resolution. Anal. Chem. 73, 647–650 (2001)
Kelleher, N.L., Senko, M.W., Siegel, M.M., McLafferty, F.W.: Unit Resolution Mass Spectra of 112 kDa Molecules with 3 Da Accuracy. J. Am. Soc. Mass Spectrom. 8, 380–383 (1997)
Wilcox, B.E., Hendrickson, C.L., Marshall, A.G.: Improved Ion Extraction from a Linear Octopole Ion Trap: SIMION Analysis and Experimental Demonstration. J. Am. Soc. Mass Spectrom. 13, 1304–1312 (2002)
Beu, S.C., Laude Jr., D.A.: Open Trapped Ion Cell Geometries for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Int. J. Mass Spectrom. Ion Processes 112, 215–230 (1992)
Hendrickson, C. L.; Beu, S. C.; Blakney, G. T.; Kaiser, N. K.; McIntosh, D. G.; Quinn, J. P.; Marshall, A. G. Optimized Cell Geometry for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Proceedings of the 57th American Society for Mass Spectrometry Conference on Mass Spectrometry and Allied Topics, 2009.
Beu, S.C., Laude Jr., D.A.: Elimination of Axial Ejection During Excitation with a Capacitively Coupled Open Trapped-Ion Cell for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 64, 177–180 (1992)
Zubarev, R.A., Kelleher, N.L., McLafferty, F.W.: Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J. Am. Chem. Soc. 120, 3265–3266 (1998)
Hakansson, K., Chalmers, M.J., Quinn, J.P., McFarland, M.A., Hendrickson, C.L., Marshall, A.G.: Combined Electron Capture and Infrared Multiphoton Dissociation for Multistage MS/MS in a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. Anal. Chem. 75, 3256–3262 (2003)
Blakney, G.T., Hendrickson, C.L., Marshall, A.G.: Predator Data Station: A Fast Data Acquisition System for Advanced FT‐ICR MS Experiments. Int. J. Mass Spectrom. (2011) doi:10.1016/j.ijms.2011.03.009
Ledford Jr., E.B., Rempel, D.L., Gross, M.L.: Space Charge Effects in Fourier Transform Mass Spectrometry. II. Mass Calibration. Anal. Chem. 56, 2744–2748 (1984)
Dey, M., Castoro, J.A., Wilkins, C.L.: Determination of Molecular Weight Distributions of Polymers by MALDI-FTMS. Anal. Chem. 67, 1575–1579 (1995)
O'Connor, P.B., Duursma, M.C., van Rooij, G.J., Heeren, R.M.A., Boon, J.J.: Correction of Time-of-Flight Shifted Polymeric Molecular Weight Distributions in Matrix-Assisted Laser Desorption/Ionization Fourier Transform Mass Spectrometry. Anal. Chem. 69, 2751–2755 (1997)
Kaiser, N.K., Skulason, G.E., Weisbrod, C.R., Wu, S., Zhang, K., Prior, D.C., Buschbach, M.A., Anderson, G.A., Bruce, J.E.: Restrained Ion Population Transfer: A Novel Ion Transfer Method for Mass Spectrometry. Rapid Commun. Mass Spectrom. 22, 1955–1964 (2008)
Caravatti, P. U.S. Patent, 1990.
Beu, S.C., Laude Jr., D.A.: Ion Trapping and Manipulation in a Tandem Time-of-Flight-Fourier Transform Mass Spectrometer. Int. J. Mass Spectrom. Ion Processes 104, 109–127 (1991)
Beu, S. C.; Hendrickson, C. L.; Marshall, A. G. SIMION Modeling of Methods to Control Time-of-Flight Dispersion of Externally Generated Ions in High Field FT-ICR MS. Proceedings of the 55th American Society for Mass Spectrometry Conference on Mass Spectrometry and Allied Topics 2005.
Limbach, P.A., Grosshans, P.B., Marshall, A.G.: Experimental Determination of the Number of Trapped Ions, Detection Limit, and Dynamic Range in Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 65, 135–140 (1993)
Cooper, H.J., Hakansson, K., Marshall, A.G.: The Role of Electron Capture Dissociation in Biomolecular Analysis. Mass Spectrom. Rev. 24, 201–222 (2005)
Tsybin, Y. O.; Hendrickson, C. L.; Beu, S. C.; Marshall, A. G. Impact of ion magnetron motion on electron capture dissociation Fourier transform ion cyclotron resonance mass spectrometry. Int. J. Mass Spectrom. 2006, 255/256, 144–149.
Wu, S., Lourette, N.M., Tolic, N., Zhao, R., Robinson, E.W., Tolmachev, A.V., Smith, R.D., Pasa-Tolic, L.: An Integrated Top-Down and Bottom-Up Strategy for Broadly Characterizing Protein Isoforms and Modifications. J. Proteome Res. 8, 1347–1357 (2009)
Parks, B.A., Jiang, L., Thomas Paul, M., Wenger Craig, D., Roth Michael, J., Boyne Michael II, T., Burke Patricia, V., Kwast Kurt, E., Kelleher Neil, L.: Top-Down Proteomics on a Chromatographic Time Scale Using Linear Ion Trap Fourier Transform Hybrid Mass Spectrometers. Anal. Chem. 79, 7984–7991 (2007)
Roth, M.J., Parks, B.A., Ferguson, J.T., Boyne II, M.T., Kelleher, N.L.: "Proteotyping": Population Proteomics of Human Leukocytes Using Top Down Mass Spectrometry. Anal. Chem. 80, 2857–2866 (2008)
Hughey, C.A., Hendrickson, C.L., Rodgers, R.P., Marshall, A.G., Qian, K.: Kendrick Mass Defect Spectrum: A Compact Visual Analysis for Ultrahigh-Resolution Broadband Mass Spectra. Anal. Chem. 73, 4676–4681 (2001)
Acknowledgments
The authors thank Dr. Jeremiah D. Tipton for help with the top-down experiments and analysis, Dr. Amy M. McKenna for help with the petroleum data collection and analysis, Daniel McIntosh for fabrication of the custom components, and Saudia Aramco (Dhahran, Saudia Arabia) for providing the crude oil sample. This work was supported by NSF Division of Materials Research through DMR-0654118 and the State of Florida.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Kaiser, N.K., Quinn, J.P., Blakney, G.T. et al. A Novel 9.4 Tesla FTICR Mass Spectrometer with Improved Sensitivity, Mass Resolution, and Mass Range. J. Am. Soc. Mass Spectrom. 22, 1343–1351 (2011). https://doi.org/10.1007/s13361-011-0141-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-011-0141-9