
Abstract
The objective of the present paper is to formulate nanostructured lipid carriers (NLCs) of a calcium channel blocker, isradipine, to enhance its oral bioavailability and prolong its antihypertensive effect apart from evaluating efficacy of the formulation in isoproterenol induced myocardial infarction in rats. Formulation was optimized using quality by design (QbD)–based approach. Three factors i.e., total lipid concentration (%), homogenization pressure (bar), and number of cycles were optimized through Box-Behnken design to estimate their effect on critical quality attributes (CQAs) viz., size (nm), % entrapment efficiency, and in vitro % drug release which were found to be 80.9 ± 1.7 nm, 83.51 ± 2.15%, and 83.3 ± 3.86% after 24 h, respectively. In vivo pharmacokinetic study indicated 4.207 and 1.907 times increase in the oral bioavailability of optimized nanostructured lipid carrier without and with cycloheximide (lymphatic transport inhibitor), respectively. Treatment with ISO (isoproterenol) significantly diverges the levels of antioxidant marker, TBARS (thiobarbituric acid), and ultrastructure of the cardiac tissue indicating significant myocardial damage. Pretreatment of nanostructured lipid carrier of isradipine (ISD-NLCs) significantly prevented the antioxidant status and ultrastructural changes in the heart. In conclusion, this study confirms that optimized NLCs can substantially improve oral bioavailability of isradipine and presents a promising strategy in the management of hypertension for longer duration of time apart from demonstrating its preclinical efficacy in cardioprotection.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hypertension, a worldwide epidemic at present, is not a disease in itself, but rather, it is an important risk factor for serious cardiovascular disorders including myocardial infarction, stroke, heart failure, and peripheral artery disease. Isradipine is a potent antihypertensive which acts on calcium channel present on cardiac and vascular tissues and inhibits the inflow of calcium resulting in vasodilatation. Unlike other calcium channel blockers, isradipine acts selectively on cardiac and skeletal muscle vasculature especially on arterial side [1]. Isradipine comes under BCS class II drug (low solubility/high permeability) which shows variable absorption pattern due to solubility limitation. Factors like low aqueous solubility, high clearance rate, hepatic first pass metabolism, intestinal metabolism, and P-glycoprotein mediated efflux of drug are the major reasons for low oral bioavailability (15–24%) of this drug [2,3,4]. Most of these problems associated with isradipine were tried to overcome by formulating NLCs of isradipine with chosen excipients. NLCs are a formulation of choice owing to the advantages offered by it, such as high drug encapsulation efficiency, biodegradability, biocompatibility, controlled release profile, and easy scale-up possibility [5]. NLCs consist of mixture of liquid lipids with solid lipids. Solid lipid nanoparticle with time tends to form perfectly crystalline lattice creating less space for drug to stay inside resulting in drug expulsion. In NLCs, formation of crystal structure is prevented due to presence of liquid lipid thus increasing the entrapment efficiency of drug and reducing the risk of gel formation during storage [6]. NLCs are formulated by lipids; drugs having high log P value are suitable candidate for NLCs formulation as loading capacity of lipophilic drugs are better in lipids as compared with hydrophilic drugs. As lymphatic uptake is possible through NLCs, therefore, hepatic first pass metabolism is circumvented [7]. The excipients having P-glycoprotein (P-gp) inhibitory effect will be used for drug which are substrate for P-gp efflux thus potentiating the effectiveness of formulation in the management of hypertension and myocardial infarction.
In earlier studies, isradipine has been found to show antioxidant property [8, 9]. Due to this action of isradipine, efficacy of isradipine in myocardial infarction (MI) was also studied. Oxidative stress can result in advancement of myocardial infarction. Such ischemic injury can be assessed by analyzing the serum level of TBARS after isradipine dosing. Cardioprotective role of isradipine can be assessed by induction of MI by isoproterenol (ISO), which is a non-selective β-agonist that shows positive chronotropic and inotropic effects at lower dose, while at higher dose, it depletes the energy reserve of cardiomyocytes.
In present work, QbD was systematically integrated to produce optimized formulation which was subsequently evaluated for its efficacy in the management of hypertension and cardioprotection.
Materials and methods
Materials
Isradipine was procured from Novartis, Basel, Switzerland. Emulcire 61 and Capryol 90 were procured from Gattefosse (Saint Priest, France). Dialysis membrane-70 (molecular weight cutoff [MWCO] = 12,000–14,000 Da), LA 393-1 MT, was purchased from HiMedia, Mumbai, India. Isoproterenol was purchased from Sigma-Aldrich, St. Louis, Missouri, USA. Poloxamer 188 was provided by BASF (India). HPLC grade acetonitrile:water was purchased from Merck India Ltd. (Mumbai, India). All other reagents used in the experiments were of AR grade.
Animals
In vivo studies were carried out as per the protocols approved by Institutional Animal Ethics Committee of Jamia Hamdard, Hamdard University, New Delhi, India (approval no. 1317). Albino Wistar rats weighing 200–250 g of either sex were taken for the experiment. Animals housed in propylene cages were kept under standard laboratory conditions maintained at temperature 25 ± 2 °C and relative humidity 55 ± 5% RH with free access to standard diet and water.
Methods
Preparation of NLCs
NLCs were prepared by high pressure homogenization method [10]. Accurate amount of solid lipid, Emulcire 61 (containing magnetic bead), and liquid lipid, Capryol 90, was weighed and transferred to a beaker. Lipid phase was melted at 60 °C. To this drug was added and stirred to obtain homogenous mass. In a separate beaker (containing magnetic bead), aqueous phase was prepared by dissolving 3% of surfactants tween 80 and poloxamer 188 in the ratio 2:1 in distilled water. Both aqueous and lipid phases were heated at same temperature 60 °C. Aqueous phase was then added to beaker containing lipid phase kept on magnetic stirrer. Hot dispersion was then subjected to high shear homogenization at 6000 rpm using RQ-127A/D, Remi, India. This hot dispersion was homogenized by high pressure homogenizer (Stansted Fluid Power Ltd. Unit 5, New Horizon, ESSEX CM19 5FN UK) at different pressure and number of cycles. Dispersion was then allowed to cool down to room temperature leading to formation of ISD-NLCs. To remove any unentrapped drug from the surface of NLC purification was performed using dialysis bag into 300 mL distilled water stirred at 300 rpm for 24 h. Distilled water was replaced after 12 h.
Optimization of NLCs as per response surface design
Box-Behnken design (BBD), a response surface methodology design, was employed to optimize formulation using Minitab 18 software (Minitab Inc., State College, PA). Table 1 represents 3 factors (total lipid concentration (%), homogenization pressure (bar), and no. of cycles, 3 levels (low, medium, and high levels of each factor) which affect dependent variables (CQAs). Each experimental run as obtained by Box-Behnken design was evaluated for their CQAs viz., size, % entrapment efficiency, and % drug release.
Selection of optimized formulation
Optimized formulation is one with desired CQAs i.e., smaller particle size, higher % EE, and higher in vitro drug release. The optimized ISD-NLCs were identified using numerical optimization method. Graphical optimization method was employed to search optimum formulation in the region of design space of overlay plot by setting the desired range of CQAs.
Evaluation of optimized formulation
Size and entrapment efficiency
Optimized ISD-NLCs were characterized on the basis of its size, polydispersity index (PDI), surface charge, and entrapment efficiency (% EE). Size of ISD-NLCs was determined by dynamic light scattering while surface charge was estimated by electrophoretic mobility of the particles under the influence of electric field using Malvern Zetasizer (Nano-ZS; Malvern instruments, Malvern, UK).
% EE of ISD-NLCs was estimated by diluting 5 mL of formulation to 10 mL with distilled water. Diluted sample was centrifuged at 40,000 rpm for 45 min using ultracentrifugation (Beckman Coulter, India Pvt. Ltd.). Supernatant was taken out and after suitable dilution with methanol drug content was determined via UV spectrophotometer. % EE were calculated using formula;
where Wtotal is the amount of drug used in preparing ISD-NLCs, and Wfree is the amount of the drug in supernatant.
In vitro release
The release studies of optimized ISD-NLCs were evaluated in simulated gastric fluid (SGF) of pH 1.2 and in simulated intestinal fluid (SIF) of pH 7.4 for 2 h and 24 h, respectively [11, 12]. Briefly, dialysis bags (molecular weight cut off 12,000 Da) containing 1 mL of formulation corresponding 1.25 mg of drug was placed in each beaker (at different pH) containing 200 mL of dissolution media (SGF and SIF) with 1% Tween 80. Stirring speed was set at 100 rpm for dissolution media maintained at temperature 37 °C on magnetic stirrer. Two milliliters of the aliquots were withdrawn at different time interval and replaced with same amount of dissolution media for maintaining constant volume. After suitable dilution, aliquots were analyzed for drug content by UV spectrophotometer.
Pharmacokinetic study
Pharmacokinetic studies in rat were investigated after oral administration of isradipine (ISD) suspension (ISD suspended in 2% sodium carboxymethyl cellulose) and ISD-NLCs at dose 12 mg/kg through 18-gauge oral feeding needle. Rats were fasted overnight with free access to water and divided into three groups (n = 4), namely, (1) ISD suspension, (2) ISD-NLCs, and (3) ISD-NLCs with cycloheximide. Cycloheximide acts as lymphatic transport inhibitor so it has been used in assessing role of NLCs in lymphatic uptake. 3 mg/kg of cycloheximide was injected to group 3 through intraperitoneal route dissolved in saline (3 mg/mL). After 1 h of injection, 12 mg/kg of ISD-NLCs were given orally to third group. 0.2 mL of blood samples were collected at different time interval (1,2,8,12 and 24 h) through tail vein in microcentrifuge tube containing EDTA. Quantification of drug in plasma was done through HPLC method.
Estimation of isradipine in plasma
Isradipine in plasma was estimated by solvent extraction and partitioning. Briefly, to 0.2 mL of rat plasma, 0.5 mL of acetonitrile was added and vortexed for a minute. To this mixture, 1 mL of ethyl acetate was added, vortexed for a minute, and then centrifuged at 4000 rpm for 10 min.
The upper organic layer was transferred to a fresh test tube and evaporated to dryness under nitrogen stream at 50 ± 2 °C. Residue was reconstituted with 0.2 mL of mobile phase, which is a combination of acetonitrile:water (50:50) and filtered through 0.25-μm membrane filter. Concentration of drug was estimated by injecting 20 µL of reconstituted sample into the HPLC.
In vitro in vivo correlation
Level A correlation representing point-to-point relationship between in vitro drug release and in vivo drug absorbed was selected. Deconvolution of in vivo plasma concentration-time data to % drug absorbed was done using Wagner Nelson’s equation (13);
where Ct is the concentration of drug at time t; \(\mathrm{AUC}0\to t\) and \(\mathrm{AUC}0\to \infty\) are area under curve between time 0 to t and 0 to infinity, respectively; and Ke is elimination rate constant with value 0.048 h−1. Successive value of % drug absorbed was calculated at different time point utilizing above equation.
Pharmacodynamic study: In vivo efficacy study in hypertensive rats
The preclinical assessment of antihypertensive activity of the developed formulation was performed on experimentally hypertensive rats. Hypertension was induced by injecting deoxycorticosterone acetate (DOCA, 5 mg/kg in corn oil) subcutaneously at every fourth day for 2 weeks [14]. Administration of DOCA elevated the systolic BP in animals. The rat was then placed in the restrainer, and the systolic blood pressure was recorded using non-invasive blood pressure system (NIBP 200A; Biopac System, Inc., Goleta, CA) based on cuff tail technique at 0, 1, 2, 4, 8, 24 h time intervals in triplicate. Following 2 weeks of treatment with DOCA, all the animals were found to be hypertensive with mean systolic blood pressure ranging 161.23–171.34 mm Hg. After 2 weeks, for antihypertensive activity studies, the animals were randomly distributed into three groups with four rats in each group. The animals in group I served as control and was given normal saline; group II and group III served as treated group and were given ISD suspension and NLCs formulation (12 mg/kg). Blood pressure of each group of the rats was recorded again to check the efficacy of the formulation.
Induction of myocardial infarction
ISO (85 mg/kg, i.p.) solutions in normal saline were injected to rats on 6th and 7th days of study to induce myocardial infarction.
Experimental protocol
Total 16 Wistar rats were used for study. After acclimatization, rats were randomly divided into 4 groups consisting of 4 rats in each group. Group I served as control and received normal saline (1 mL) orally for 7 days. Group II served as toxic group and received normal saline (1 mL) orally for 7 day s and ISO (85 mg/kg, i.p.) on 6th and 7th days. Group III and IV served as test group and received isradipine and ISD-NLCs, respectively, both at same dose of 12 mg/kg/day orally for 7 days and ISO (85 mg/kg, i.p.) on 6th and 7th days.
Twenty-four hours after the last dose, blood from each group was collected through tail vein of rat and centrifuged to separate serum, which were stored at −20 °C ± 5 °C for the biochemical analysis. Animals were then euthanized by CO2 anesthesia, and hearts were immediately removed, washed with normal saline. Small piece of heart was preserved in 2.5% glutaraldehyde and 2% paraformaldehyde solution in 0.1 M sodium phosphate buffer (pH 7.2) for transmission electron microscopy. Remaining heart was kept at −20 °C for biochemical estimations.
Biochemical estimation
In myocardial tissue, the level of lipid peroxide was determined as nmol malondialdehyde (MDA) per mg of protein. Procedure as earlier reported by Ohkawa et al. (1979) to estimate level of lipid peroxide was used without any modification [15].
Briefly, to 0.2 mL of 10% heart homogenate, 0.2 mL of 8.1% sodium dodecyl sulfate, 1.5 mL of 20% acetic acid adjusted to pH 3.5, and 1.5 mL of 0.8% aqueous solution of thiobarbituric acid were added. Volume was adjusted to 4 mL by adding distilled water which was then heated in oil bath for 1 h at 95 °C. Mixture was cooled and to it 1 mL distilled water, and 5 mL mixture of n-butanol and pyridine (15:1, v/v) was added and shaken vigorously. The content was centrifuged at 4000 rpm for 10 min. The supernatant (organic layer) was taken out, and its absorbance was read at 532 nm. 1,1,3,3-tetramethoxypropane (TMP) as external standard to generate standard plot from where the concentration of MDA as nmol in heart tissue can be obtained.
Transmission electron microscopy studies
Small pieces (1–1.5 mm) of animal myocardial tissue of left ventricle were taken and rinsed several time with 0.1 M phosphate buffer of pH 7.2. Samples were then immediately fixed with 2.5% glutaraldehyde and 2% paraformaldehyde solution in 0.1 M sodium phosphate buffer (pH 7.2) and stored at 4 °C for 12 h. Samples were stained with 2% uranyl acetate and 2% lead acetate and evaluated under transmission electron microscope (TEM) at All India Institute of Medical Sciences and Research (AIIMS) New Delhi, India [16].
Statistical analysis
All the results were expressed as mean ± standard deviation. One-way analysis of variance (ANOVA) was used to compare groups followed by Dennett’s t-test. In all the cases, p < 0.05 was considered statistically significant.
Results
Formulation and analysis of NLCs as per response surface design
Three levels three factors experimental design with 15 experimental trials are shown in Table 2. Each CQA was found to follow quadratic model as given by the equation in Table 3. As per ANOVA, each CQA represented R2 > 97%, indicating goodness of fit of quadratic model.
Supplementary data Figs. S1(A) and S1(B) demonstrate that, as the total lipid concentration increases, a curvilinear increase in particle size can be observed at low level and high level of both pressure and no. of cycles indicating that they are not interacting with fluctuation in the value of total lipids. Supplementary data Fig. S1(C) shows inverted umbrella shape curve indicating as homogenization pressure increases till intermediate value size decreases markedly. Further increase in pressure causes increase in the size of particle. At low value of pressure as the no. of cycles increases, size decreases till mid value of no. of cycles while further increase causes increase in particle size. At low pressure but at high value of pressure as the no. of cycles increases, size increases linearly. But at high value of pressure, size increases almost linearly as no. of cycles is increased.
Supplementary data Fig. S1(D) indicates as the concentration of total lipid increases significant increase in the % EE results till mid value of lipid concentration at both low and high levels of no. of cycles. Further increase in total lipid concentration causes decrease in % EE. As the homogenization pressure increases, % EE decreases linearly. Supplementary data Fig. S1(E) indicates umbrella shape 3D curve revealing the same effect of total lipids on % EE. Supplementary data Fig. S1(F) depicts marked decrease in the value of % EE with increasing pressure irrespective of no. of cycles revealing no interaction of pressure with no. of cycles.
Supplementary data Fig. S1(G) depicts curvilinear decrease in % drug release as concentration of total lipids increases. The value of % drug release was taken at the time point of 24 h throughout the study. This trend is irrespective of magnitude of pressure applied revealing no interaction of pressure with concentration of total lipids. Supplementary data Fig. S1(H) indicates increase in % drug release as the no. of cycles increases at low concentration of total lipid. But at high concentration of total lipids, increase in no. of cycles causes decrease in % drug release. Supplementary data Fig. S1(I) is umbrella shape 3D curve. As the pressure increases, % drug release increases to maximum while further increase in pressure after intermediate value while further increase in pressure causes decrease in % drug release. As the no. of cycles increases till its mid value, % drug release increases after that decrease in its value is observed independent of the magnitude of homogenization pressure.
Selection of optimized formulation
Response optimizer was used to find out the optimum formulation by setting the desired goal. No constrain was put on factors like total lipid (2.86%) and homogenization pressure (500 bar) while nos. of cycles were hold at 6. Supplementary data Table S1 indicates the composition of the factors to obtain desired response with composite desirability of 0.854. Best formulation was selected from design space in the overlay plot after narrowing down the constraints as depicted in Fig. 1.

Overlay plot indicating design space for ISD-NLC
Validation of optimization studies
Five checkpoint formulations were prepared, and their observed responses were compared with predicted responses as given by Minitab 18.0. Supplementary data Fig. S2 illustrates that correlation plots prepared between observed and predicted responses demonstrate high R2 value which was found to be greater than 0.96 in each case indicating excellent goodness of fit.
Evaluation of optimized formulation
Size, zeta potential, and entrapment efficiency
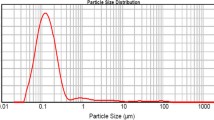
Particle size of ISD-NLCs was found to be 80.9 ± 1.7 nm with PDI 0.251 ± 0.034 as shown in Supplementary Data Fig. S3(A). Zeta potential (Supplementary Data Fig. S3(B)) and entrapment efficiency of the optimized formulation was found to be −15.8 mV and 83.51 ± 2.15% respectively.
In vitro release
28.8 ± 2.94% of drug was released in SGF in 2 h while 83.3 ± 3.86% of drug was released in SIF for 24 h (Fig. 2). Almost one-third of the drug was released from SGF in first 2 h indicating quick drug release followed by sustained release of drug for 24 h in SIF. % drug release in 24 h from ISD-NLC was significantly higher than that of isradipine drug suspension (p < 0.001) at pH 7.4.

In vitro release study of ISD-NLC and drug suspension using dialysis method at pH 1.2 and 7.4. Data expressed as mean ± SD, n = 3
Pharmacokinetic study
Plasma drug concentration-time profiles of ISD suspension and ISD-NLCs with and without cycloheximide are shown in Fig. 3. Difference in the plasma concentration of ISD-NLCs as compared with ISD-NLCS with cycloheximide and ISD suspension at each time point was significant (p < 0.01). Pharmacokinetic parameters exhibited by the formulation (ISD-NLCs) showed remarkable increase in oral bioavailability as evident from Table 4. Oral bioavailability of ISD-NLCS improved by 4.2-fold and 1.9-fold in contrast to ISD suspension and ISD-NLCs with cycloheximide, respectively. There was significant difference (p < 0.01) in Cmax and t1/2 of ISD-NLCs (6987.64 ng/mL/h and 14.31 h) as compared with ISD-NLCs with cycloheximide (1893.66 ng/mL/h and 11.84 h) and ISD suspension (2354.25 ng/mL/h and 8.52 h).

Plasma drug concentration–time profile of orally administered ISD suspension, ISD-NLC and ISD-NLC with cycloheximide (Cx) at the dose of 12 mg/kg. Data expressed as mean ± SD, n = 4
In vitro in vivo correlation
On plotting in vitro % drug release and in vivo % drug absorbed as represented in Fig. 4, linear correlation with R2 of 0.9645 was obtained thereby indicating that in vitro test can be used to predict in vivo performance of the ISD-NLCs.

In vitro in vivo correlation of ISD-NLC using Wagner Nelson method
Pharmacodynamic study
In vivo antihypertensive activity was performed for 24-h period. All the animals of group I was found to be hypertensive with mean systolic blood pressure between 161.23 and 171.34 mm Hg. Significant reduction in mean systolic blood pressure (p < 0.001) was found with treatment groups II and III i.e., 119.35 ± 18.81 mm Hg and 123.74 ± 8.4 mm Hg, respectively, after 1 h of oral administration as compared with group I (Fig. 5). Further, ISD-NLCs were found to show mean systolic blood pressure less than 130 mm Hg for the entire 24-h period. ISD suspension showed maximum reduction in blood pressure of 131.26 ± 14.08 mm Hg for 4 h, and after that, there was elevation in blood pressure. Results suggested that developed formulation of ISD-NLCs improves the oral delivery of ISD.

In vivo antihypertensive activity of animals orally administered with normal saline, ISD suspension and ISD-NLC at the dose of 12 mg/kg. Values expressed in mean ± SD (n = 4)
Biochemical estimation
Figure 6 represents the effect of different groups on the level of thiobarbituric acid-reactive substance (TBARS). By determining content of malondialdehyde (MDA), level of TBARS was evaluated. Group II (toxic group, ISO-treated) showed significant (p < 0.001) increase in the level of TBARS as compared with control group I (untreated, control). Group III (toxic group treated with ISD) and group IV (toxic group treated with ISD-NLC) represent significant decrease in the level of TBARS (p < 0.01) as compared with toxic group II. When group IV was compared with group III, significant decrease in the level TBARS of ISD-NLCs was observed at p < 0.05.

Effect of ISD suspension and ISD-NLC on antioxidant parameters in myocardial tissues during isoproterenol-induced myocardial infarction in rats. Data expressed as mean ± SD (n = 4 animals per group). Vehicle control: normal saline (0.5 mL/kg, i.p.); ISO: toxic group (85 mg/kg, i.p.); ISD + ISO: ISD (12 mg/kg, oral) + ISO (85 mg/kg, i.p.); ISD-NLC + ISO: ISD-NLC (12 mg/kg, oral) + ISO (85 mg/kg, i.p.)
Effect on the ultrastructure of cardiac tissue
Figure 7 shows effect of different treated groups on the ultrastructure of left ventricle. Group I: (normal control) group showed normal architecture of heart mitochondria and myofilament. Groups II: (toxic group) treated with Isoproterenol (ISO; 85 mg/kg, i.p.) showed swollen heart mitochondria with the ruptured mitochondrial cell membrane, the band was totally damaged, protein got hampered, and cell death was seen. Group III: group treated with ISD-NLCS (12 mg/kg, i.p) was showing one intact nucleus. Mitochondria are normal along with good appearance of myofilaments of the heart; cristae are normal.

Transmission electron microscopic study of the ventricle tissue of rat heart. Group I (normal control) shows normal mitochondria with normal myofilament with visible Z-band. Group II (toxic group) treated with ISO (85 mg/kg, i.p.) results in cell death with totally damaged Z-band. Group III treated with ISD-NLC shows restoration of myofilament band. Cell membrane is normal with intact nucleus
Discussion
Accommodation of the drug in the mixture of solid lipid and liquid lipid is higher due to their spatial dissimilarity and increased imperfection in highly ordered crystal lattice structure of solid lipid. This imperfection also decreases time-dependent % drug expulsion from NLCs [17]. Similar phenomenon occurred increasing isradipine solubility in the mixture of Emulcire 61 and Capryol 90 in contrast to their individual counterpart.
Three factors were selected finally to prepare optimized formulation through BBD. Effect of each factor on CQAs was studied by response surface plots. Curvilinear increase in the size with increase in total lipid concentration could be due to unavailability of adequate surfactant to emulsify the system which resulted in agglomeration of the particles [18]. Increase in particle size with increase in number of cycles could be attributed by particle aggregation. Increase in homogenization pressure resulted in smaller particle size as high kinetic energy is obtained by particle due to pressure leading to disruption in size. Excess pressure and number of cycles caused particle aggregation which led to increase in particle size [19].
Till intermediate concentration of total lipid, % EE increases because more amount of lipid was available to solubilize the drug. In other words, extra space becomes available to entrap more drug. The concentration of surfactant throughout the experiment was kept constant. As the concentration of lipid increases, there is increased possibility of reduction in emulsifying efficiency of surfactant and thus hindering the nanoparticles formation apart from aggregation of nanoparticles. This instability could be the reason of decrease in EE% [20]. Increase in number of cycles after intermediate level decreases % EE because, when the particle breaks due to high kinetic energy, then there is increased possibility of drug to get out of the particles. As the homogenization pressure was increased from minimum to maximum level, % EE decreased to significant level as high force disrupted the particle leading to drug loss [19].
Due to increase in the total lipid concentration, significant decrease in the drug release was observed due to increase in the viscosity of the formulation which hinders drug release [21]. Both, excess pressure and number of cycles, resulted in decrease in % drug release. This could be attributed by increase in particle agglomeration which hindered release of drug. But minimum to intermediate increase in pressure showed different trend, which was increase in % drug release. Smaller size of particle as obtained by intermediate supply of pressure could be the reason, as smaller size has large effective surface area.
Amount of drug release from SIF was higher as compared with SGF. This result could be due to presence of bile salts which acts as natural wetting agent and promotes dissolution and release of the drug [11]. In first 2 h, drug release was quick which is necessary for immediate therapeutic action. Quick drug release was followed by sustained drug release for 24-h period to maintain therapeutic effect of drug for longer duration of time. This sustained release profile could be attributed due to presence of drug well inside oil droplets which is surrounded by solid lipid matrix [12].
Increased oral bioavailability of ISD-NLCs as compared with ISD suspension could be attributed due to smaller size of the NLCs and pre-enterocyte solubilization and absorption of the drug in lipid nanoformulation. In addition, it was found that surfactants like Tween 80 and Poloxamer 188 act as P-gp efflux inhibitor, which could have contributed in marked enhancement of drug level in plasma [6, 22]. Besides, in small size of the NLCs (< 100 nm), ISD-NLCs protects drug from hepatic metabolism thus increasing concentration of drug in plasma [23].
Lipid-based carrier gets transported to lymphatic circulation by two different ways either by transcytosis uptake through M-cells which covers the Peyer’s patches of small intestine or by increasing chylomicron production in the enterocytes which covers the NLCs surface and is secreted into lymph [24]. In cycloheximide treated rats, lower concentration of drug in plasma as compared with untreated rats reveals the role of lymphatic uptake of drug from oral lipid-based carrier (NLCs). Cycloheximide acts as lymphatic transport inhibitor by hindering the production of chylomicrons in enterocyte and thus transport of lipid-based carrier into lymphatic circulation [25]. As seen in Fig. 5, maintenance of low systolic blood pressure for 24 h is effectively achieved by ISD-NLCs. This could be attributed by the sustained release action of the ISD-NLCs which has maintained the therapeutic range for 24-h duration. Release of drug from NLCs generally involves three processes. First is dissolution of the drug present at the surface of NLCs which results in immediate action of the drug in lowering blood pressure. Second is diffusion of the drug from the NLCs due to formation of the concentration gradient between drug present in the NLCs and dissolution media (Intestinal media). Third is degradation of the lipid surface of NLCs resulting in slow release of the drug from the core of NLCs resulting in sustained action of the drug and thus maintenance of low blood pressure for 24-h duration [26].
Oxidative stress is one of the factors involved in cardiomyopathy as evaluated by ISO treatment. However, exact pathogenesis of cardiomyopathy is not clear. ISD apart from hypertension has been found to show its effect in Parkinson disease due to its neuroprotective role [27]. ISD has also been found to have antioxidant effect [8]. Concentration of MDA increases when rats are treated with ISO. In higher concentration, MDA being the marker of cardiac damage indicates the decrease in the antioxidants enzyme activity [28]. ISD and ISD-NLCs at doses 12 mg/kg reversed the increased levels of MDA thereby decreased lipid peroxidation. Effect of ISD-NLCs on the level of lipid peroxide was found to be significantly better (p < 0.01) than ISD suspension (p < 0.05) when compared with the ISO group thus showing efficacy of ISD-NLCs over ISD suspension.
The transmission electron micrographs of the mitochondria of the heart in ISO group indicated damaged band protein with swelling in mitochondria which resulted in cell death. Such type of mitochondria is characteristic feature of myocardial ischemia. Rats pre-treated with ISD-NLCs in ISO-treated groups showed restoration of myofilament bands with normal cell structure.
Conclusion
ISD-NLCs were successfully optimized to achieve sustained release with higher release rate indicating that dosing frequency of the drug can be decreased. Efficacy of drug in NLCs formulation enhanced due to increase in bioavailability which suggested that low dose of drug in its NLCs formulation is possible thus avoiding adverse drug effect. ISD-NLCs showed better protection in ISO induced MI as evident from the estimation of TBARS and TEM study. Considering these results, it can be concluded that ISD-NLCs can be effectively utilized in the management of hypertension and is potentially effective against ISO induced MI. The current study is a good example of formulation which provides therapeutic value for the patients who develops hypertension and issue like MI, as the formulation demonstrated its efficacy in both the cases.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Veld AJM. The place of isradipine in the treatment of hypertension. Am J Hypertens. 1991;4:96S-102S.
Verger ML, Fluckiger L, Kim Y, Hoffman M, Maincent P. Preparation and characterization of nanoparticles containing an antihypertensive agent. Eur J Pharm Biopharm. 1998;46:137–43.
Tran TT, Tran PH, Nguyen MNU, Tran KTM, Pham MN, Tran PC, Vo TV. Amorphous isradipinenanosuspension by the sonoprecipitation method. Int J Pharm. 2014;474(1–2):146–50.
Ramasahayam B, Eedara BB, Kandadi P, Jukanti R, Bandari S. Development of isradipine loaded self-nano emulsifying powders for improved oral delivery: in vitro and in vivo evaluation. Drug Dev Ind Pharm. 2015;41(5):753–63.
Venugopal V, Kumar KJ, Muralidharan S, Parasuraman S, Raj PV, Kumar KV. Optimization and in vivo evaluation of isradipine nanoparticles using Box-Behnken design surface response methodology. Open Nan. 2016;1:1–15.
Fathi HA, Allam A, Elsabahy M, Fetih G, El-Badry M. Nanostructured lipid carriers for improved oral delivery and prolonged antihyperlipidemic effect of simvastatin. Colloids Surf B Biointerfaces. 2017;162:236–45.
Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6:231–48.
Lupo E, Locher R, Weisser B, Vetter W. In vitro antioxidant activity of calcium antagonists against LDL oxidation compared with α-tocopherol. Biochembiophys res commun. 1994;203(3):1803–8.
Godfraind T. Antioxidant effects and the therapeutic mode of action of calcium channel blockers in hypertension and atherosclerosis. Philos Trans R SocLond B BiolSci. 2005;360:2259–72.
Amasya G, Aksu B, Badilli U, Onay-Besikci A, Tarimci N. QbD guided early pharmaceutical development study: production of lipid nanoparticles by high pressure homogenization for skin cancer treatment. Int J Pharm. 2019;563:110–21.
Shrestha N, Bouttefeux O, Vanvarenberg K, Lundquist P, Cunarro J, Tovar S, Khodus G, Andersson E, Keita AV, Dieguez CG, Artursson P, Preat V, Beloqui A. Stimulation of GLP-1 secretion and delivery of GLP-1 agonists via nanostructured lipid carriers. Nanoscale. 2018;10(2):603–13.
Shete H, Patravale V. Long chain lipid based tamoxifen NLCS. Part I: preformulation studies, formulation development and physicochemical characterization. Int J Pharm. 2013;454:573–584.
D’Souza S, Faraj JA, Giovagnoli S, DeLuca PP. IVIVC from long acting olanzapine microsphere. Int J Biomat. 2014;1–12.
Qadri GR, Ahad A, Aqil M. Invasomes of isradipine for enhanced transdermal delivery against hypertension: formulation, characterization, and in vivo pharmacodynamic study. Artif cells nanomedbiotechnol. 2017;45(1):139–45.
Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95(2):351–8.
Pogodina M, Shornikova YS, Chentsov luS. Electron microscopy description of cardiomyocytes from the left ventricle of rat heart after apoptosis induction by isoproterenol. Biol Bull. 2006;33(1):19–29.
Kaithwas V, Dora CP, Kushwah V, Jain S. Nanostructured lipid carriers of olmesartanmedoxomilwithenhanced oral bioavailability. Colloids Surf B Biointerfaces. 2017;154:10–20.
Alam T, Khan S, Gaba B, Haider MF, Baboota S, Ali J. Adaptation of quality by design-based development of isradipinenanostructurede lipid carrier and its evaluation for in vitro gut permeation and in vivo solubilization fate. J Pharm Sci. 2018;107(11):2914–26.
Severino P, Santana MHA, Souto EB. Optimizing SLN and NLCS by 22 full factorial design: effect of homogenization technique. Mater SciEng C. 2012;32:1375–9.
Subedi RK, Kang KW, Choi HK. Preparation and characterization of solid lipid nanoparticles loaded with doxorubicin. Eur J Pharm Sci. 2009;37:508–13.
Shah B, Khunt D, Bhatt H, Misra M, Padh H. Intranasal delivery of venlafaxine loaded nanostructured lipid carrier: risk assessment and QbD based optimization. J Drug DelivSciTechnol. 2016;33:37–50.
Jawahar N, Hingarh PK, Arun R, Selvaraj J, Anbarasan A, Sathianarayanan S, Nagaraju G. Enhanced oral bioavailability of an antipsychotic drug through nanostructured lipid carriers. Int J BiolMacromol. 2018;110:269–75.
Attari Z, Bhandari A, Jagadish PC, Lewis S. Enhanced ex vivo intestinal absorption of olmesartanmedoxomilnanosuspension: preparation by combinative technology. Saudi Pharm J. 2016;24:57–63.
Tiwari R, Pathak K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int Pharm. 2011;415:232–43.
Khan S, Shaharyar M, Fazil M, Hassan MQ, Baboota S, Ali J. Tacrolimus-loaded nanostructured lipid carriers for oral delivery-in vivo bioavailability enhancement. Eur J Pharm Biopharm. 2016;109:149–57.
Luan J, Zheng F, Yang X, Yu A, Zhai G. Nanostructured lipid carriers for oral delivery of baicalin: in vitro and in vivo evaluation. Colloids and Surfaces A: PhysicochemEng Aspects. 2015;466:154–9.
Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol Dis. 2011;43:364–71.
Ansari MA, Iqubal A, Ekbbal R, Haque SE. Effects of nimodipine, vinpocetine and their combination on isoproterenol induced myocardial infarction in rats. Biomed Pharmacother. 2019;109:1372–80.
Funding
This project was financially assisted by the Hamdard National Foundation (HNF), New Delhi.
Author information
Authors and Affiliations
Contributions
Conceptualization: Tausif Alam; methodology: Tausif Alam, Mohd Asif Ansari; formal analysis and investigation: Tausif Alam; writing—original draft preparation: Tausif Alam; writing—review and editing: Javed Ali, Sanjula Baboota; funding acquisition: Hamdard; resources: Javed Ali; supervision: Javed Ali, Sanjula Baboota.
Corresponding author
Ethics declarations
Consent for publication
I, Dr. Javed Ali, corresponding author of this research article on behalf of all the authors (Mr. Tausif Alam, Mr. Asif Ansari, Dr. Sanjula Baboota) hereby confirm that this article is original, does not infringe on any copyright, is not under consideration by any other journal, and has not been previously published. Ethical approval for conducting experiment on animals has been sought and obtained as necessary.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Alam, T., Ansari, M.A., Baboota, S. et al. Nanostructured lipid carriers of isradipine for effective management of hypertension and isoproterenol induced myocardial infarction. Drug Deliv. and Transl. Res. 12, 577–588 (2022). https://doi.org/10.1007/s13346-021-00958-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-021-00958-x