
Abstract
Fibers of poly(L-lactic acid) (PLLA)/polyhydroxybutyrate (PHB) with different concentrations of the drug dipyridamole (DPD) were prepared using solvent-free melt electrospinning to obtain a polymeric drug delivery system. The electrospun fibers were morphologically, structurally, thermally, and dynamically characterized. Crazes that resemble lotus root crevices were interestingly observed in the 7:3 PLLA/PHB fibers with 1% DPD. The crystallinity of PLLA slightly decreased as PHB was incorporated, and the addition of DPD significantly reduced the melting temperature of the composite. The interactions between PLLA and PHB mainly occurred at a proportion of 7:3, and drug encapsulation in the fibers was verified. The kinetic profiles of drug release demonstrated the predominant multiple patterns involving a diffusional stage in the short-term mode of release and kinetic process related to the hydrolysis of the biopolymers. Furthermore, the dynamic behavior of the polymer molecules was evaluated based on the segmental mobility using probe electron spin resonance spectroscopy. The segmental mobility in the amorphous fraction of PLLA decreased with increasing PLLA content. The 9:1 PLLA/PHB system was more resistant to polymer hydrolysis than to the 7:3 system and the rate of diffusion transport was approximately two times higher for the 7:3 PLLA/PHB fibers than for the 9:1 PLLA/PHB fibers.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Controlled drug delivery systems offer numerous advantages, including improved therapeutic effects, reduced toxicity, and increased patient compliance and convenience, over conventional dosage forms [1]. In such systems, synthetic, natural, and hybrid polymer materials are used as the drug vehicles to enhance the efficiency of the system [2, 3]. Polyhydroxybutyrate (PHB) is a biocompatible, nontoxic, and biodegradable polymer obtained from natural source that is suitable for biomedical applications [4]. This polymer is one of the most promising biomedical materials because of its appropriate biocompatibility and controlled biodegradation. PHB has a high degree of crystallinity because of its perfect stereoregularity and high purity. However, the excessive brittleness, poor processability, and poor thermal stability of PHB limit its potential applications as a pristine material [5]. Multicomponent fibers have attracted increasing attention because new properties can be obtained by combining different materials. We combined PHB with other biopolymers to create a drug delivery system to investigate the diffusion transport kinetics of drugs. Poly(L-lactic acid) (PLLA) was chosen because it has been widely used in various biomedical applications [6,7,8] based on its biodegradability, biocompatibility, and good mechanical properties.
Unresolved problems for polymeric drug delivery systems include their low efficiency for preparing nano- and microparticles or vesicles, and their low efficiency for drug delivery [9]. Electrospinning is a simple, low-cost, and versatile process for preparing a wide range of polymeric microfibers and nanofibers in a controllable manner [10]. Moreover, this technique is very convenient because the fiber can be coated onto any surface with nonwoven or aligned fibers. Electrospinning can be generally divided into two types of methods: solution electrospinning and melt electrospinning. Solution electrospinning is usually used to prepare nanofibers because it uses simple equipment and produces thinner fibers than melt electrospinning. However, many of the residual solvents in the fibers are toxic to cells and tissues [11]. In contrast, melt electrospinning provides a solvent-free process to fabricate polymer fibers using a safe, eco-friendly method. Thus, the disadvantages of solution electrospinning, such as expensive solvent recovery, risk of solvent explosion, and residual solvent toxicity, are not applicable. These features are becoming increasingly important in the pharmaceutical industry.
The chosen drug, dipyridamole (DPD), is an antithrombotic and antithrombogenic drug. It can reduce smooth muscle cell proliferation and promote vascular endothelial cell proliferation. In addition, DPD has been employed with acetylsalicylic acid for secondary stroke prevention because of its antithrombotic effect [12]. DPD had been blended with different polymers and then electrospun into a biodegradable fibrous scaffold or other forms for drug delivery [13,14,15]. However, the toxic solvent residue in the fibers cannot be eliminated because it must be used to dissolve the polymer. Thus, in this study, solvent-free melt electrospinning was applied to prepare novel drug delivery systems.
Controlled drug release from nondegradable polymer systems occurs through a diffusion mechanism [16,17,18], while degradable polyesters, such as PLLA and PHB, with unstable chemical bonds, can be affected by hydrolytic reactions, specifically through the mechanism of end group autocatalysis [19]. In this connection, despite the impressive technological achievements in the creation of ultrathin fibers, an extremely limited amount of work [20,21,22] has been devoted to diffusion and hydrolytic problems that emerge in fibrillar materials during drug delivery. These problems require comprehensive experimental and theoretical examinations. To solve the coherent diffusion-kinetic problems, measurements of controlled drug release from the macroscopic fibrillar membranes (the mats) under various intrinsic conditions must be executed.
In the present work, fiber membranes with various proportions of PLLA/PHB were produced by melt electrospinning. The developed materials were morphologically, structurally, thermally characterized. PLLA/PHB fiber membranes without any toxic solvents and containing different amounts of DPD were prepared to assess the polymeric drug delivery system. The influence of DPD on the fiber diameter and the structural and thermal properties were characterized. Further, drug encapsulation in the fibers was confirmed. The dynamic behavior of the polymer molecules was evaluated by their segmental mobility and drug diffusion, which is the most important process for controlled drug release.
Experimental
Materials
PHB (1001MD, BASF) and PLLA particles (Mn = 100,000; Zhejiang Hisun Biomaterials Co., Ltd., China) were dried at 60 °C for 6 h in a vacuum oven before the experiments. DPD (Mw = 504.63; > 98.0%) was purchased from Beijing Inoke Technology Co. Ltd., China.
Processing of the blends
Blends of PLLA and PHB at different PLLA/PHB ratios of 9:1, 8:2, 7:3, and 6:4 by weight were mixed in a Haake polylab torque rheometer at 190 °C for 6 min at a constant rotor speed of 80 rpm. The PLLA/PHB ratios of 9:1 and 7:3 with 1% and 5 DPD by weight are the same condition except for the temperature at 170 °C.
Melt electrospinning
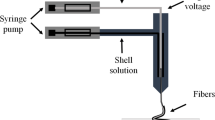
The melt electrospinning devices used are the same as those in our previous studies [23, 24]. The collector covered with aluminum foil was connected to the positive of the power, and the spinneret was grounded. Samples were prepared using a normal power supply (DW-P503-2ACDE; Dong Wen high-voltage power supply (Tianjin) Co., Ltd., China). First, several electrospinning processing conditions were tried to produce the electrospinning fiber of PLLA and the ratio of 9:1. The different positive voltages, applied cylinder temperature, and spinning distance were investigated for the formation of a stable Taylor cone to obtain uniform and continuous fiber. Because of the addition of the DPD, the applied temperatures could be decreased notably, and it was fixed to 170 °C. The DPD acts as the plasticizer in melt electrospinning and decreases polymer-polymer interaction to make segments of polymer move at low temperature [25]. Thus, the melt electrospinning was carried out at 220 °C without DPD and 170 °C with DPD, the applied voltage was 35 kV, and the distance between collector and spinneret was set at 7 cm. The schematic diagram of the experimental devices and as the spun fiber of 7:3 with 1% DPD is shown in Fig. 1.

Schematic diagram of the experimental devices and the as-spun PLLA/PHB fibers of 7:3 with 1% DPD
Characterization
Electrospun fiber membranes were coated with gold and their morphology was characterized using scanning electron microscopy (SEM, Hitachi S4700, Japan) with an accelerating voltage of 20 kV. The average fiber diameter and its size distribution were calculated using SEM images by ImageJ software. Over approximately 20 fibers of each sample were randomly selected from the SEM image, and each fiber was measured at five different locations. The crystalline phases of the electrospun fibers were performed using X-ray diffraction (XRD) (D8, Bruker, Germany) in the 2θ range 5°–90° with Cu Kα radiation (λ = 1.5406 Å), with a voltage and current which were 40 kV and 40 mA, respectively. The equipment resolution and the scan speed were 0.02° and 0.1 s per step, respectively. Thermophysical characteristics and crystallization behavior were obtained by differential scanning calorimetry (DSC, PerkinElmer Pyris l) with the samples being heated from 0 to 200 °C at 10 °C min−1 under a nitrogen atmosphere (20 mL min−1). The glass transition temperature (T g ), cold crystallization temperature (T c ), melting temperature (T m ), cold crystallization enthalpy (ΔH c ), and melting enthalpy (ΔH m ) were obtained from the first heating, and the degree of crystallinity X C (%) was calculated using Eq. 1 [26]:
ΔH f is the melting heat associated with pure crystalline PLLA (93 J g−1) [27] and 1/W PLLA is the proportion of PLLA in the blend. Infrared measurements (FTIR) were performed at room temperature with a Thermo Electron 8700 apparatus in transmission mode from 4000 to 500 cm−1. FTIR spectra were collected after 10 scans with a resolution of 4 cm−1. The measurements were performed with potassium bromide pellets (KBr). Segmental mobility of PLLA and PHB in the blend fibers was studied by probe electron spin resonance spectroscopy (ESR) method. X-band EPR spectra were registered on an automated EPR-V spectrometer (Semenov Institute of Chemical Physics, Moscow RF.). A stable nitroxide radical TEMPO (2,2,6,6-tetramethylpyperidin-1-oxil) was used as a probe. The radical was introduced into the fibers from the gas phase at 40 °C. The radical concentration in the polymer was not above 10−3 mol/L. The experimental spectra of the spin probe in the region of slow motions (τ > 10−10 s) were analyzed within the model of isotropic Brownian rotation using a program Budil et al. described [28]. The spectra were modeled using the following main values of the g-tensor and the hyperfine coupling tensor of the radical: gxx = 2.0096, gyy = 2.0066, gzz = 2.0025, Ахх = 7.0G, Ayy = 5.0 G, and Azz = 35.0 G. The value Azz was determined experimentally from the EPR spectra of the nitroxide radial in the polymer at − 216 °C; it was almost equal to the values Timofeev et al. reported [29]. The correlation time of probe rotation, τ, were determined from the ESR spectra via the equation [30]:
where ∆Н+ is the width of spectrum component located in a weak field and I+/I– is the ratio of component intensities in weak and strong fields of the spectrum, respectively. The statistical error of τ measurements is equal to ± 5. The kinetics of DPD release was studied with Beckman DU-65 UV spectrophotometer (USA). In kinetic measurements, the fiber sample was immersed in an aqueous medium and the optical density of DPD samples was determined with a periodic sampling. The interval of sampling depended on the fiber composition and, accordingly, on the rate of drug release; it was from 1 to 30 min. The experiments lasted from several decades of minutes to several hours. In the DPD UV spectra, there are two characteristic peaks at λ = 410 nm and a more intense peak at λ = 292 nm with an extinction coefficient of 31, 260 L/(mol cm) which was used for the drug release measurement.
Results and discussion
Fiber membrane morphology
SEM micrographs of electrospun fibers in the membranes are shown in Fig. 2a–h. The fibers of the as-spun PLLA, PHB, and their blends (PLLA/PHB = 9:1 and 7:3) without DPD have smooth surfaces and relatively uniform fiber diameter distributions without any specific thickening, such as beads or spindle-like units. In contrast to the drug-unloaded fibers, the composite fibers with a PLLA/PHB ratio of 7:3 and 1% DPD have rough surfaces and nonuniform diameters within a single fiber. In this case, their morphology includes crazes that resemble lotus root crevices; the crazes are apparently disunited, but remain connected to the nanofibrils. Analogous morphologies were discovered for the polymer samples under tensile testing [31, 32]. However, we obtained similar structures without any intentional stretching during melt electrospinning.

Morphology of the electrospun fiber membranes of a pure PLLA and for PLLA/PHB ratios, b 9:1, c 8:2, d 7:3, e 6:4, f pure PHB, and 7:3 with g 1 and h 5% of DPD concentration; i the corresponding average fiber diameters
In the present study, the crazes in the fibers are not evenly spaced along the fiber axis, and they are not present for some single filaments. In the absence of external mechanical stress and under identical electrospinning conditions for the nonloaded and drug-loaded samples, these fiber defects developed only in the presence of DPD and, hence, are directly related to the DPD molecule impact upon the fiber morphology and the segmental mobility of polymer molecules (see the ESR results, in the “ESR probe spectroscopy of PLA/PHB electrospun mats” section). Generally, these effects are likely related to (1) a slight polymer phase separation between the 7:3 polymer blends and DPD [33], (2) electrically driven jet instabilities during electrospinning (non-axisymmetric instability and two axisymmetric instabilities), which form a multipoint local stress concentration on the jet [32] and further expand the fluctuation, and (3) a decrease in the entanglement of molecular chains form the addition of DPD, which contributes to the motion of the molecular chains. A further increase in the DPD concentration to 5 wt% causes the PLLA/PHB fiber disintegration. Figure 2h shows that the samples are composed of short, crescent-like units that are very likely caused by jet discontinuity during electrospinning.
All electrospun fiber formulations and the corresponding average diameters are summarized in Table 1. The fiber diameter decreases as the PHB content increases in the composite fibers; for 7:3 PLLA/PHB fibers, the fiber diameter reaches minimum value of 18.8 ± 6.0 μm. The smallest diameter is observed for pristine PHB, as shown in Fig. 2i. DPD embedded in the polymer system decreases the mean fiber diameter to 10.7 ± 2.5 μm and simultaneously creates a narrower diameter distribution. The reduced fiber thickness from encapsulated DPD was previously described [34]; the authors proposed that ionic forms of DPD participate in the electrostatic interactions during fiber electrospinning. Simultaneously, the authors observed a surface tension reduction in the presence of DPD; this effect could cause fiber bending with the following discontinuity of the polymer jet [35]. The coherent consequence of this effect is observed in Fig. 2h for the 7:3 compositions with 5% DPD loading.
Fourier transformed infrared spectroscopy
FTIR spectra of the PLLA, PHB, and PLLA/PHB membranes are shown in Fig. 3 band associated with the C-CH3 stretching vibrations of PLLA appearing at 1044 cm−1, and its intensity decreases with PHB addition. The band at 1080 cm−1 is ascribed to the stretching vibrations of the C–O–C groups. In neat PHB, the band at 1271 cm−1 is ascribed to stretching vibrations of the C–O–C groups, which decrease with PLLA addition [36]. At 1384 cm−1, the CH3 asymmetric deformation band appears, and its intensity increases with higher amounts of PHB [37]. In the spectrum of PLLA, the typical band at 1752 cm−1 is related to the crystalline carbonyl C=O vibration [38]. The amorphous carbonyl C=O vibration of PLLA is centered at 1745 cm−1, but it is very weak and cannot be clearly identified [37]. In the spectrum of pure PHB, the bands at 1718 and 1733 cm−1 are attributed to the stretching vibrations of crystalline carbonyl C=O groups, and a small shoulder at 1750 cm−1 contributes to the amorphous carbonyl C=O vibration [38, 39]. For the PLLA/PHB blends, the carbonyl stretching bands of PLLA and PHB show different intensities with increasing of PHB content. For the 6:4 blend, the band at 1722 cm−1 is ascribed to stretching of the crystalline carbonyl group, and the band near 1733 cm−1 becomes narrower and overlaps with the amorphous carbonyl group of PLLA. In the 7:3 blend, two distinct carbonyl groups are observed: the band at 1752 cm−1 corresponds to the amorphous carbonyl vibration of PLLA, and the band at 1716 cm−1 corresponds to crystalline carbonyl stretching of PHB. However, for the 9:1 blend, the carbonyl band at 1745 cm−1 broadens and shows a small shoulder near 1718 cm−1. The different peak position in the blends is associated with a transesterification reaction between PLLA and PHB that forms intermolecular hydrogen [40].

FTIR spectra of electrospun membranes with PLLA/PHB blends
The FTIR spectra show the typical characteristic vibrational bands of DPD at 1534 cm−1 corresponding to the C=N (ring) stretching vibration (Fig. 4) [41]. A new absorption band at 1582 cm−1 corresponding to the C=O stretching vibration appears, which may be associated with the formation of a carbonyl group during the melt electrospinning [42]. In this way, the presence of DPD was confirmed in the membranes after melt electrospinning.

FTIR spectra of DPD and the ratio of 7:3 blend electrospun membranes with 0, 1, and 5% DPD
Differential scanning calorimetry
The heating processes of PLLA, PHB, and their blends with and without DPD are observed in the DSC scans (Fig. 5a). Pure PLLA has a glass transition temperature (T g ) of ~ 61 °C and a sharp melting peak of ~ 173 °C. For pure PHB, a small melting peak of 150 °C is observed, and the glass transition is not discernible. Pure PHB shows a lower melting point than the general because it has a poor thermal stability in the melt and thermal degradation begins at the melting point [43].

DSC scans (a) during the heating processes, and XRD (b) of PLLA, PHB, and PLLA/PHB blends with 0, 1, and 5% DPD
For the PLLA/PHB blends, a glass transition is observed at ~ 57 °C for the PLLA component, and the glass transition temperature minorly varies with changing composition. The crystallization peak of PLLA appears at 108 °C, while that of PHB does not appear clearly. The DSC thermal properties are summarized in Table 2. The crystallization peak for the 9:1 blend appears at 86 °C, and the crystallization temperature decreases as the PLLA/PHB ratio changes from 9:1 to 6:4. The decreasing crystallization temperature indicates a faster crystallization rate of PLLA in the blend than pure PLLA based on the ability of PHB to recrystallize the PLLA matrix [37, 40]. The melt temperature slightly decreases with the increasing PHB content. A small exothermal peak at 159 °C appears below the melting point; it may originate from the imperfect crystals that form during the electrospinning process and is affected by the heating rate [44,45,46]. The melting peak of all blends and PLLA shows a small significant shoulder, which is ascribed to the presence of two crystal forms and recrystallization effects during the heating process [47]. The crystallinity of PLLA generally increases as PHB is incorporated using the solution method [26, 37]; however, in this study, the crystallinity slightly decreases, which can be explained by two considerations: (1) melt electrospinning generally forms amorphous or semi-crystalline structures [48] and (2) molten polymers quenched at room temperature do not immediately crystallize [49]. Since the degree of tension has not reached the critical value of crystallization because of the rapid temperature drop, molecular chains in the melt are drawn by the electric force and solidify at the low temperature. Therefore, most of the molecular chains have a certain degree of orientation along the tensile direction, but the crystallinity is relatively low.
The T g of PLLA/PHB with 1% DPD is slightly higher than that of PLLA/PHB. However, the T g of PLLA/PHB with 5% DPD is significantly lower than that of PLLA/PHB. Although no significant differences in the crystallization temperature are observed between the blend with 1% DPD and neat PLLA/PHB, the crystallization temperature of the blend with 5% DPD is increased. For the melting behavior, the addition of DPD significantly reduces the melting temperature of the blend by redistributing of the crystals [50]. The melting peak becomes narrower, and the small shoulder that was observed for the neat PLLA/PHB blend is not observed for the blend with 1% DPD; this indicates that the formation of more stable crystals is promoted by adding DPD.
X-ray diffraction
The crystalline structure of the electrospun fibers was investigated by XRD, and the diffraction patterns are shown in Fig. 5b. An amorphous, broad band is typically observed for the pure PLLA sample [51]. The weaker peak located at 2θ = 31.0° is associated with the β crystal, and the broad dispersion peaks at 16.6° and 19.0° are related to the (103) and (203) reflections, respectively, of the α-form crystal of PLLA [52, 53]. The diffraction pattern for the electrospun film contains five peaks at 17.4°, 20.3°, 23.2°, 25.5°, and 29.6°, corresponding to the (110), (021), (111), (121), and (200) reflections of PHB, respectively [54]. The presence of crystalline PHB does not produce significant signals or increase the crystallinity of the PLLA matrix, in agreement with the slight decrease in the degree of crystallinity calculated from the DSC results. In the 6:4 blend, weak peaks are observed at 2θ = 17.4°, 23.2°, and 29.6°. These findings suggest that the crystal structure of PHB in the 6:4 electrospun membrane is altered; the crystal growth rate of PLLA is slower than that of PHB, which creates interactions between PLLA and PHB [54]. The 7:3 blend with 1% DPD shows a slight increase in the intensity of the peaks at 2θ = 17.4°, 20.3°, 23.2°, and 29.6° with respect to those for the pure 7:3 blend; this may be attributed to the high dispersion of DPD, which contributed to crystal formation.
ESR probe spectroscopy of PLA/PHB electrospun mats
The last four sections presented structural, morphological, and thermal information, which showed that special features exist for the PLLA/PHB blend fibers fabricated by melt electrospinning. The next two sections will describe the dynamic behavior of polymer molecules, namely their segmental mobility, as evaluated using a probe ESR method and DPD diffusion measurements, which are the most important processes for controlled drug release, especially for its short-term development. The molecular structural features, morphology, and physicochemical characteristics provide the necessary conditions, while the kinetics of diffusive transport in combination with the hydrolysis rate provide sufficient conditions for drug release in biodegradable fibers.
A series of ESR spectra for the (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) radical probe encapsulated in the PLLA/PHB systems (microfibers and microparticles) are shown in Fig. 6. The shapes of all ESR curves indicate clearly that each inherent spectrum manifests the superposition of two individual spectra that belong to the two populations of radicals with two different correlation times, τ 1 and τ 2. Here, the intrinsic correlation time, τ 1, designates the status of the radical in the dense amorphous fields with a slow rotation mobility, while τ 2 designates fast radical rotation in the less dense amorphous fields of the fibers. The latter fields are designated as the “soft” fields. The existence of two TEMPO populations in the amorphous phase of PHB and PLLA with distinctive rotation frequencies indicates the heterogeneous structure of the intercrystalline areas of the biopolymers. The state of the intercrystalline fraction could be approximated by a two-mode model, which was previously proposed for several semicrystalline polymers, such as polyalcanoates, polylactides, and poly(ethylene terephthalate) [55, 56].

Probe ESR spectra of TEMPO radicals embedded in PLLA/PHB fibrillar systems: (1) 9:1/5%; (2) 7:3/5%; (3) 9:1/1%; (4) 7:3/1%
Quantitative analysis of the ESR spectra was performed by evaluating the effective correlation times in accordance with the above Formula (2). Additionally, the ratios of the intensities of the two first low-field peaks, I+ 1 and I+ 2, belong to slow and fast rotations, respectively (see Fig. 7); the activation energy of the radical rotational mobility, E a , was also measured. All characteristics are summarized in Table 3.

ESR spectrum deconvolution (#2 in Fig. 6). a The fast and slow components of ESR spectrum after deconvolution. b The comparison of the real initial spectrum (1) with the spectrum evaluated as superposition (2) of two different modes shown in a
The differences in the peak intensities of the ESR spectra show that the effective correlation time, τ C, in the 9:1 PLLA/PHB fibers exceeds the same characteristic of the 7:3 PLLA/PHB fibers, which also indicates the decline in segmental mobility in the amorphous fraction of PLLA with an increase in the PLLA content. The concentration effect is quite explicable, especially considering that the T g of PLLA more than three dozen degree exceeds the T g of PHB. Hence, at room temperature, the former is in a glassy state where segmental mobility is essentially hindered, while the latter has the limited features of an elastic polymer; despite the high crystallinity, its segmental mobility is more intense. The relative standard deviations of the correlation times are 6%, the intensity ratio is 10%, and the activation energy of the probe mobility is 5%.
A comparison of the values for the I+ 1/I+ 2 ratio showed that the partition coefficients of the radicals in the dense and soft fields of the amorphous fraction of the polymer do not differ much. Nevertheless, the perfect fibrils show slightly higher intensity ratios (see Table 3) than the distorted PLLA/PHB systems (see Fig. 2h). This result points out the denser structural organization in the inter-crystalline area in the well-formulated fibrillars. Here, it is appropriate to mention the work [57] where it showed an analogous small discrepancy between the meaning of the I+ 1/I+ 2 ratio for the ultrathin fibrils and the cold-rolled films of the pristine PHB.
In Table 3, the important characteristics of radical mobility are presented as energy activation values calculated from the Arrhenius equation in the coordinates log (τ C) vs. 1/T K. The Arrhenius plots for microparticles are not monotonic and show specific breakpoints that reflect a sharp change in the slope of the corresponding straight lines at ~ 57 and ~ 39 °C for the systems containing 5% DPD and with polymer ratios of 9:1 and 7:3, respectively. The decrease in the rotational activation energy is likely related to acceleration of the rotation probe mobility near the glassy state transition in PLLA, as in the dominant component (T g = 58 °C for the 9:1 ratio, see Table 2). For the 7:3 system, PHB impacts the total mobility of the radical; thus, we could treat this mobility transition as the superposition of the segmental dynamics of both biopolymers. In any case, this temperature effect demands the following investigations. Finally, all these transitions are observed for the mats formed with microparticles only at higher DPD contents (Fig. 8).

Semilogarithmic dependence of radical rotational mobility on reciprocal absolute temperature for PLLA/PHB systems. The arrows show the breakpoints expressed in Celsius degrees
Impact of diffusion upon controlled drug release
Figure 9a demonstrates the typical kinetic profiles of drug release from the PLLA/PHB fiber mats with DPD loading and different ratios of the polymer components. As for drug release from the PHB films [58], the kinetic profiles have two inherent sections characterized by different shapes for the mat: linear and nonlinear. In accordance with the proposed diffusion-kinetic model, the initial nonlinear section of each curve principally reflects the drug diffusion process, whereas the linear section corresponds to the kinetic process of partial loss of polymer weight because of the onset of the hydrolytic decomposition of PLLA and PHB ester groups. During hydrolysis, the encapsulated drug moves into aqueous surroundings, not only by a diffusion mechanism but also by surface degradation.

a Kinetic profiles of drug release from PLLA/PHB mats. b Diffusion impact upon the drug controlled release (DPD). Kinetic curves reflect drug diffusivity in the polymer phase
For the macroscopic fibrillar membranes (the mats) formed by single filaments, by analogy with the monolithic nonporous film, the diffusion-kinetic equation can be written to reflect both diffusion and hydrolysis simultaneously as
where С d and G d are the total concentration of the drug passing from the polymer into the external aqueous volume, V, and the concentration of the drug that can diffuse into the polymer, respectively; D eff is the effective diffusion coefficient, which is independent of coordinates and time, and k h is the constant of the zero-order hydrolytic reaction of PLLA and PHB ester groups on the filament surface.
The PLLA/PHB mats consist of disordered entangled fibers, and D eff is determined for these mats by two consecutive processes: the drug diffusion mobility in the inherent fiber volume (D f) and drug transport in the pores consisting of interfibrous space and filled by solvent (D w). By describing the two-stage drug transport as a sequence of proper diffusion in the fiber and subsequent transfer in the interfibrous space, that is, modeling the diffusion mode of release as a two-layered medium in accordance with the Crank simplification [59], D eff could be presented as
where X f and L w are the average characteristic sizes of the drug diffusion path length in the proper fiber and the interfibrous space, respectively.
For the cylindrical fibers, X f is the fiber diameter, while for L w, in accordance with the Mackie-Mears equation [60], the correction for increasing the drug diffusion path based on its tortuosity was chosen as
where φ f is the volume fraction of polymer fibers. This correction was previously used to describe the drug diffusion in the PHB magnetic composites with the magnetite nanoparticles embedded forming the extended aggregates [61].
The diffusion equation for the cylindrical fibers loaded with uniformly distributed drug was advanced by Crank [59]:
where r is the coordinate of the radial diffusion; symbol G d, as in Eq. (3), denotes the concentration of the mobile fraction of the drug in the cylindrical fiber with the corresponding constant diffusion coefficient D f, and X f is the average diameter of the fiber. The initial and symmetrical boundary conditions corresponding to the drug desorption from the cylindrical fibers are the following: G f = G f 0 at t = 0 (at the initial time) and G f = 0 at r = R (at the fiber/solution interface).
In accordance with the research of J. Siepmann et al. [62], the solution of differential Eq. (6) makes it possible to obtain the dependence of the cumulative amount of the desorbed drug (M t ) on the time of release (t):
where M ∞ is the limiting value of M t under the condition t → ∞.
For Eqs. (6) and (7), which both reflect drug transfer through the surface of the side walls and in a cylinder, the cylindrical fiber is at least five times longer than its radius [63]. This mathematical requirement is fully fulfilled for electrospun fibers of practically infinite length. Additionally, the latter equation is valid when M t /M ∞ ≤ 0.4. The combination of Eqs. (3) and (7) gives the final expression for drug release from cylindrical fibers that are simultaneously subjected to hydrolysis and diffusion:
where k c = k h − [4D f/X f 2]. The positive sign in the equation shows that the condition, k h > [4D f/X f 2], is fulfilled for the PLLA/PHB fibers, which is confirmed in Fig. 9b. For a relatively short exposure time (~ 8 h) and given the S-mechanism of PLA and PHB hydrolysis under short-term controlled release [64], no changes are observed in the viscosity, molecular weight, or crystallinity. The experimental curves are consistent with that provided by Eq. (8), which provides a method to evaluate the drug diffusivity in the diffusion coordinates: M t /M ∞ ≈ t 1/2. Table 4 lists the drug release characteristics of the studied systems, including the diffusion coefficients (D f) from Eq. (8). The low values of D f satisfactorily agree with the same low values for diffusion mobility of DPD in the PHB microparticle [65].
The penultimate column in Table 4 shows the values of k h, which were calculated from the curves presented in Fig. 9b using the last member of Eq. (8). The 9:1 PLLA/PHB system is more resistant to polymer hydrolysis than the 7:3 system. The partial incompatibility of PLLA and PHB could increase the microphase separation surface of the single filaments, making them more accessible to attack by the hydrolytic agent.
The right column of Table 4 shows the mobile fraction of the drug encapsulated in the microfibrils, which are capable of diffusion. Simultaneous collation of the effective DPD diffusivities in combination with the values of the DPD mobile fractions shows that the rate of diffusion transport is approximately twice as high for the fibers with less PLLA (i.e., 7:3) than for those in the 9:1 system. This effect is associated with the essential drop in fiber crystallinity, which is shown in Table 2. In accordance with the two-phase model for the amorphous areas of polymers, which was confirmed using ESR (see “ESR probe spectroscopy of PLA/PHB electrospun mats”), the rest of the drug molecules are tightly encapsulated in the amorphous, dense areas and do not participate in diffusion transport. This immobilized fraction is only capable of drug release because of the surface destruction in accordance with a zero-order reaction. DPD release provides multiple biofunctions for biodegradable nanofibrous films, which show promise for use in biodegradable, small-diameter vascular grafts and other blood-contact implants and are candidates for drug delivery systems for sustained delivery of pharmaceutical agents for applications related to cardiovascular disease.
Conclusions
In this study, the fiber membranes of PLLA/PHB blends with different concentrations of DPD were fabricated using solvent-free melt electrospinning to create controlled-release systems. The fibers without DPD had a smooth surface and a relatively uniform fiber diameter distribution. The fibers with DPD showed a rough surface and nonuniform diameters within a single fiber. The fibers with DPD included crazes that resembled lotus root crevices. The DSC results suggested that the crystallinity of PLLA slightly decreased with PHB incorporation; this was further confirmed using XRD. The addition of DPD significantly reduced the melting temperature of the blend. FTIR spectroscopy showed a transesterification reaction between PLLA and PHB, and confirmed that the drug was immobilized on the fibers.
Furthermore, the dynamic behavior of the polymer molecules was evaluated by segmental mobility and drug diffusion. In the blend fibers, the segmental mobility of PLLA and PHB declined with increasing PLLA content. The values of I+ 1/I+ 2 indicate a denser structural organization of the inter-crystalline area in the fibrils. The decrease in the rotation activation energy is likely related to acceleration of the rotation probe mobility near the glassy-state transition. The consideration of drug release under short-term conditions showed that these nanofibers can be characterized by the diffusion mode of a sustained drug release profile. The 9:1 PLLA/PHB system is more resistant to polymer hydrolysis than the 7:3 system. The simultaneous collation of the effective DPD diffusivities in combination with the DPD mobile fractions shows that the rate of diffusion transport is approximately two times higher for the 7:3 PLLA/PHB fibers than for the 9:1 PLLA/PHB fibers.
References
Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric systems for controlled drug release. Chem Rev. 1999;99(11):3181–98. https://doi.org/10.1021/cr940351u.
Kong L, Ziegler GR. Fabrication of pure starch fibers by electrospinning. Food Hydrocoll. 2014;36:20–5. https://doi.org/10.1016/j.foodhyd.2013.08.021.
Langer R. Drug delivery and targeting. Nature. 1998;392(6679 Suppl):5–10.
Ali AQ, Kannan TP, Ahmad A, Samsudin AR. In vitro genotoxicity tests for polyhydroxybutyrate—a synthetic biomaterial. Toxicol in Vitro. 2008;22(1):57–67. https://doi.org/10.1016/j.tiv.2007.08.001.
You J, Chiu H, Don T. Spherulitic morphology and crystallization kinetics of melt-miscible blends of poly(3-hydroxybutyrate) with low molecular weight poly(ethylene oxide). Polymer. 2003;44(15):4355–62. https://doi.org/10.1016/S0032-3861(03)00348-3.
Santos D, Silva DM, Gomes PS, Fernandes MH, Santos JD, Sencadas V. Multifunctional PLLA-ceramic fiber membranes for bone regeneration applications. J Colloid Interface Sci. 2017;504:101–10. https://doi.org/10.1016/j.jcis.2017.05.032.
Oyama HT, Tanishima D, Ogawa R. Biologically safe poly(l-lactic acid) blends with tunable degradation rate: microstructure, degradation mechanism, and mechanical properties. Biomacromolecules. 2017;18(4):1281–92. https://doi.org/10.1021/acs.biomac.7b00016.
Badia JD, Reig-Rodrigo P, Teruel-Juanes R, Kittikorn T, Strömberg E, Ek M, et al. Effect of sisal and hydrothermal aging on the dielectric behavior of polylactide/sisal biocomposites. Compos Sci Technol. 2017;149(8):1–10. https://doi.org/10.1016/j.compscitech.2017.05.026.
Jing Z, XY X, Chen XS, et al. Biodegradable electrospun fibers for drug delivery. J Control Release. 2003;92(3):227–31.
Xie G, Wang Y, Han X, Gong Y, Wang J, Zhang J, et al. Pulsed electric fields on poly-L-(lactic acid) melt electrospun fibers. Ind Eng Chem Res. 2016;55(26):7116–23. https://doi.org/10.1021/acs.iecr.6b00958.
Dalton PD, Klinkhammer K, Salber J, Klee D, Möller M. Direct in vitro electrospinning with polymer melts. Biomacromolecules. 2006;7(3):686–90. https://doi.org/10.1021/bm050777q.
Diener HC, Cunha L, Forbes C, Sivenius J, Smets P, Lowenthal A. European Stroke Prevention Study 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke1. J Neurol Sci. 1996;143(1–2):1–13. https://doi.org/10.1016/S0022-510X(96)00308-5.
D'Ilario L, Francolini I, Martinelli A, Piozzi A. Dipyridamole-loaded poly(L-lactide) single crystals as drug delivery systems. Macromol Rapid Commun. 2007;28(18–19):1900–4. https://doi.org/10.1002/marc.200700224.
Repanas A, Bader A, Klett A, Ngezahayo A, Glasmacher B. The effect of dipyridamole embedded in a drug delivery system made by electrospun nanofibers on aortic endothelial cells. J Drug Deliv Sci Technol. 2016;35:343–52. https://doi.org/10.1016/j.jddst.2016.08.011.
Punnakitikashem P, Truong D, Menon JU, Nguyen KT, Hong Y. Electrospun biodegradable elastic polyurethane scaffolds with dipyridamole release for small diameter vascular grafts. Acta Biomater. 2014;10(11):4618–28. https://doi.org/10.1016/j.actbio.2014.07.031.
Hassani Besheli N, Mottaghitalab F, Eslami M, Gholami M, Kundu SC, Kaplan DL, et al. Sustainable release of vancomycin from silk fibroin nanoparticles for treating severe bone infection in rat tibia osteomyelitis model. ACS Appl Mater Interfaces. 2017;9(6):5128–38. https://doi.org/10.1021/acsami.6b14912.
Harnoy AJ, Buzhor M, Tirosh E, Shaharabani R, Beck R, Amir RJ. Modular synthetic approach for adjusting the disassembly rates of enzyme-responsive polymeric micelles. Biomacromolecules. 2017;18(4):1218–28. https://doi.org/10.1021/acs.biomac.6b01906.
Martinelli A, D’Ilario L, Francolini I, et al. Water state effect on drug release from an antibiotic loaded polyurethane matrix containing albumin nanoparticles. Int J Pharm. 2011;407(1–2):197–206. https://doi.org/10.1016/j.ijpharm.2011.01.029.
Wang Y, Pan J, Han X, Sinka C, Ding L. A phenomenological model for the degradation of biodegradable polymers. Biomaterials. 2008;29(23):3393–401. https://doi.org/10.1016/j.biomaterials.2008.04.042.
Keshavarz P, Ayatollahi S, Fathikalajahi J. Mathematical modeling of gas–liquid membrane contactors using random distribution of fibers. J Membr Sci. 2008;325(1):98–108. https://doi.org/10.1016/j.memsci.2008.07.025.
Hopfenberg HB. In: Paul DR, Harris FW, editors. Controlled release polymeric formulations. Washington, DC: American Chemical Society; 1976. p. 26.
Azwa ZN, Yousif BF, Manalo AC, Karunasena W. A review on the degradability of polymeric composites based on natural fibres. Mater Des. 2013;47:424–42. https://doi.org/10.1016/j.matdes.2012.11.025.
Li X, Liu Y, Peng H, Ma X, Fong H. Effects of hot airflow on macromolecular orientation and crystallinity of melt electrospun poly(L-lactic acid) fibers. Mater Lett. 2016;176:194–8. https://doi.org/10.1016/j.matlet.2016.04.070.
Liu Y, Li X, Ramakrishna S. Melt electrospinning in a parallel electric field. J Polym Sci Polym Phys. 2014;52(14):946–52. https://doi.org/10.1002/polb.23511.
Nagy ZK, Balogh A, Dravavoelgyi G, et al. Solvent-free melt electrospinning for preparation of fast dissolving drug delivery system and comparison with solvent-based electrospun and melt extruded systems. J Pharm Sci-US. 2013;102(2):508–17. https://doi.org/10.1002/jps.23374.
Arrieta MP, López J, López D, Kenny JM, Peponi L. Development of flexible materials based on plasticized electrospun PLA–PHB blends: structural, thermal, mechanical and disintegration properties. Eur Polym J. 2015;73:433–46. https://doi.org/10.1016/j.eurpolymj.2015.10.036.
Repanas A, Glasmacher B. Dipyridamole embedded in polycaprolactone fibers prepared by coaxial electrospinning as a novel drug delivery system. J Drug Deliv Sci Technol. 2015;29:132–42. https://doi.org/10.1016/j.jddst.2015.07.001.
Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg–Marquardt algorithm. J Magn Reson. 1996;120(2):155–89. https://doi.org/10.1006/jmra.1996.0113.
Timofeev VP, Misharin AY, Tkachev YV. Simulation of EPR spectra of the radical TEMPO in water-lipid systems in different microwave ranges. Biophysics. 2011;56(3):420–32.
Buchachenko AL, Vasserman AM. In stable radicals. Moscow: Khimiya; 1973.
Asran AS, Seydewitz V, Michler GH. Micromechanical properties and ductile behavior of electrospun polystyrene nanofibers. J Appl Polym Sci. 2012;125(3):1663–73. https://doi.org/10.1002/app.34847.
Yoshioka T, Dersch R, Greiner A, Tsuji M, Schaper AK. Highly oriented crystalline PE nanofibrils produced by electric-field-induced stretching of electrospun wet fibers. Macromol Mater Eng. 2010;295(12):1082–9. https://doi.org/10.1002/mame.201000207.
Liu Y, Liu Y, Lee J, et al. Ultrafine formation of optically transparent polyacrylonitrile/polyacrylic acid nanofibre fibrils via electrospinning at high relative humidity. Compos Sci Technol. 2015;117:404–9. https://doi.org/10.1016/j.compscitech.2015.07.017.
Ling X, Spruiell JE. Analysis of the complex thermal behavior of poly(L-lactic acid) film. II. Samples crystallized from the melt. J Polym Sci Polym Phys. 2006;44(23):3378–91. https://doi.org/10.1002/polb.20987.
Zeng J, Yang L, Liang Q, Zhang X, Guan H, Xu X, et al. Influence of the drug compatibility with polymer solution on the release kinetics of electrospun fiber formulation. J Control Release. 2005;105(1–2):43–51. https://doi.org/10.1016/j.jconrel.2005.02.024.
Hu Y, Sato H, Zhang J, Noda I, Ozaki Y. Crystallization behavior of poly(l-lactic acid) affected by the addition of a small amount of poly(3-hydroxybutyrate). Polymer. 2008;49(19):4204–10. https://doi.org/10.1016/j.polymer.2008.07.031.
Furukawa T, Sato H, Murakami R, Zhang J, Duan YX, Noda I, et al. Structure, dispersibility, and crystallinity of poly(hydroxybutyrate)/poly(l-lactic acid) blends studied by FT-IR microspectroscopy and differential scanning calorimetry. Macromolecules. 2005;38(15):6445–54. https://doi.org/10.1021/ma0504668.
Sato H, Murakami R, Padermshoke A, Hirose F, Senda K, Noda I, et al. Infrared spectroscopy studies of CH••O hydrogen bondings and thermal behavior of biodegradable poly(hydroxyalkanoate). Macromolecules. 2004;37(19):7203–13. https://doi.org/10.1021/ma049117o.
Padermshoke A, Katsumoto Y, Sato H, Ekgasit S, Noda I, Ozaki Y. Melting behavior of poly(3-hydroxybutyrate) investigated by two-dimensional infrared correlation spectroscopy. Spectrochim Acta A. 2005;61(4):541–50. https://doi.org/10.1016/j.saa.2004.05.004.
Zhang M, Thomas NL. Blending polylactic acid with polyhydroxybutyrate: the effect on thermal, mechanical, and biodegradation properties. Adv Polym Technol. 2011;30(2):67–79. https://doi.org/10.1002/adv.20235.
Borges CPF, Tabak M. Spectroscopic studies of dipyridamole derivatives in homogeneous solutions: effects of solution composition on the electronic absorption and emission. Spectrochim Acta A. 1994;50(6):1047–56. https://doi.org/10.1016/0584-8539(94)80026-X.
Silva OM, Miguel Agustinho SC, Guzzi Plepis AMD, et al. On the thermal decomposition of dipyridamole: thermogravimetric, differential scanning calorimetric and spectroscopic studies. Spectrosc Lett. 2006;39(2):145–61. https://doi.org/10.1080/00387010500531126.
Kunioka M. Y. Thermal degradation of microbial copolyesters: poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and poly(3-hydroxybutyrate-co-4-hydroxybutyrate). Macromolecules. 1990;23(1):26–31.
Ling X, Spruiell JE. Analysis of the complex thermal behavior of poly(L-lactic acid) film. I. Samples crystallized from the glassy state. J Polym Sci Polym Phys. 2006;44(22):3200–14. https://doi.org/10.1002/polb.20972.
Zhang J, Duan Y, Sato H, Tsuji H, Noda I, Yan S, et al. Crystal modifications and thermal behavior of poly(l-lactic acid) revealed by infrared spectroscopy. Macromolecules. 2005;38(19):8012–21. https://doi.org/10.1021/ma051232r.
Zong XH, Kim K, Fang DF, et al. Structure and process relationship of electrospun bioabsorbable nanofiber membranes. Polymer. 2002;43(16):4403–12. https://doi.org/10.1016/S0032-3861(02)00275-6.
Monticelli O, Bocchini S, Gardella L, Cavallo D, Cebe P, Germelli G. Impact of synthetic talc on PLLA electrospun fibers. Eur Polym J. 2013;49(9):2572–83. https://doi.org/10.1016/j.eurpolymj.2013.05.017.
Brown TD, Daltona PD, Hutmacher DW. Melt electrospinning today: an opportune time for an emerging polymer process. Prog Polym Sci. 2016;56:116–66. https://doi.org/10.1016/j.progpolymsci.2016.01.001.
Furuhashi Y, Imamura Y, Jikihara Y, Yamane H. Higher order structures and mechanical properties of bacterial homo poly(3-hydroxybutyrate) fibers prepared by cold-drawing and annealing processes. Polymer. 2004;45(16):5703–12. https://doi.org/10.1016/j.polymer.2004.05.069.
Karpova SG, Ol'Khov AA, Shilkina NG, et al. Influence of drug on the structure and segmental mobility of poly(3-hydroxybutyrate) ultrafine fibers. Polym Sci Ser A. 2017;59(1):58–66. https://doi.org/10.1134/S0965545X17010060.
Channuan W, Siripitayananon J, Molloy R, Sriyai M, Davis FJ, Mitchell GR. The structure of crystallisable copolymers of l-lactide, ε-caprolactone and glycolide. Polymer. 2005;46(17):6411–28. https://doi.org/10.1016/j.polymer.2005.04.097.
El-Hadi AM, Mohan SD, Davis FJ, et al. Enhancing the crystallization and orientation of electrospinning poly (lactic acid) (PLLA) by combining with additives. J Polym Res. 2014;21(12):1–12.
Nisha SK, Asha SK. Random copolyesters containing perylene bisimide: flexible films and fluorescent fibers. ACS Appl Mater Interfaces. 2014;6(15):12457–66. https://doi.org/10.1021/am502248u.
Abdelwahab MA, Flynn A, Chiou B, Chiellini E, et al. Thermal, mechanical and morphological characterization of plasticized PLA–PHB blends. Polym Degrad Stab. 2012;97(9):1822–8. https://doi.org/10.1016/j.polymdegradstab.2012.05.036.
Kamaev PP, Aliev II, Iordanskii AL, Wasserman AM. Molecular dynamics of the spin probes in dry and wet poly(3-hydroxybutyrate) films with different morphology. Polymer. 2001;42(2):515–20. https://doi.org/10.1016/S0032-3861(00)00339-6.
Di Lorenzo ML, Gazzano M, Righetti MC. The role of the rigid amorphous fraction on cold crystallization of poly(3-hydroxybutyrate). Macromolecules. 2012;45(14):5684–91. https://doi.org/10.1021/ma3010907.
Staroverova O, Karpova S, Iordanskii A, et al. Comparative dynamic characteristics of electrospun ultrathin fibers and films based on poly(3-hydroxybutyrate). Chem Chem Technol. 2016;10(2):151–8. https://doi.org/10.23939/chcht10.02.151.
Iordanskii AL, Rogovina SZ, Kosenko RY, Ivantsova EL, Prut EV. Development of a biodegradable polyhydroxybutyrate-chitosan-rifampicin composition for controlled transport of biologically active compounds. Dokl Phys Chem. 2010;431(2):60–2. https://doi.org/10.1134/S0012501610040020.
Crank J. The mathematics of diffusion. 2nd ed. Oxford: Clarendon Press; 1975.
Mackie JS, Meares P. The diffusion of electrolytes in a cation-exchange resin membrane. I. Theoretical. Proc R Soc Lond. 1955;232(1191):498–509. https://doi.org/10.1098/rspa.1955.0234.
Bychkova AV, Iordanskii AL, Kovarski AL, Sorokina ON, Kosenko RY, Markin VS, et al. Magnetic and transport properties of magneto-anisotropic nanocomposites for controlled drug delivery. Nanotechnol Russ. 2015;10(3–4):325–35. https://doi.org/10.1134/S199507801502007X.
Siepmann J, Siepmann F. Mathematical modeling of drug delivery. Int J Pharm. 2008;364(2):328–43. https://doi.org/10.1016/j.ijpharm.2008.09.004.
Iordanskii AL, Olkhov AA, Karpova SG, et al. Influence of the structure and morphology of ultrathin poly(3-hydroxybutyrate) fibers on the diffusion kinetics and transport of drugs. Polym Sci Ser A. 2017;59(3):352–62. https://doi.org/10.1134/S0965545X17030075.
Bonartsev AP, Boskhomodgiev AP, Iordanskii AL, Bonartseva GA, Rebrov AV, Makhina TK, et al. Hydrolytic degradation of poly(3-hydroxybutyrate), polylactide and their derivatives: kinetics, crystallinity, and surface morphology. Mol Cryst Liq Cryst. 2012;556(1):288–300. https://doi.org/10.1080/15421406.2012.635982.
Bonartsev AP, Livshits VA, Makhina TA, Myshkina VL, Bonartseva GA, Iordanskii AL. Controlled release profiles of dipyridamole from biodegradable microspheres on the base of poly(3-hydroxybutyrate). Express Polym Lett. 2007;1(12):797–803. https://doi.org/10.3144/expresspolymlett.2007.110.
Acknowledgements
The authors gratefully acknowledge Dr. Karpova IBCP (ESR spectra measuring) and Dr. R.Yu. Kosenko ICP and Dr. E.L. Kucherenko ICP (drug release performances).
Funding
The paper was financially supported by the National Natural Science Foundation of China (Grant Number 21374008). This research was partly supported by National Grant of OKHNM RAN 2016 (the Head Prof A.R. Khokhlov).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Highlights
Solvent-free electrospinning was applied for preparing drug delivery composites.
Segmental mobility of PLLA declines with the raise of amorphous content.
PLLA/PHB systems at 9:1 ratio are more resistant to polymer hydrolysis.
Fibers with crazes that resemble the lotus root crevice were found.
Rights and permissions
About this article

Cite this article
Cao, K., Liu, Y., Olkhov, A.A. et al. PLLA-PHB fiber membranes obtained by solvent-free electrospinning for short-time drug delivery. Drug Deliv. and Transl. Res. 8, 291–302 (2018). https://doi.org/10.1007/s13346-017-0463-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-017-0463-7