
Abstract
Fulminant type 1 diabetes (FT1D) is characterized by a relatively low HbA1c level at the onset, despite the abrupt occurrence of marked hyperglycemia with ketosis or ketoacidosis. The initial symptoms/findings are flu-like, absence of islet-associated autoantibodies, and a drastic decrease in β-cells and α-cells, which strongly suggest the involvement of a viral infection. In fact, we successfully demonstrated that a FT1D-like phenotype can be reproduced in encephalomyocarditis virus-induced diabetes murine model. However, there is a discussion on the possible involvement of autoimmunity rather than viral infection as the underlying cause of FT1D. For example, HLA-DRB1*04:05, a susceptible antigen of type 1A diabetes, is reportedly associated with FT1D in Japan. Moreover, anti-glutamic acid decarboxylase antibody is reportedly detected in ~ 5% of the patients. Additionally, half of the patients with anti-programmed cell death-1 therapy-related type 1 diabetes fulfilled the criteria of the disease. These findings suggest that islet-associated autoimmunity can partially contribute to the development of FT1D. Furthermore, using nonobese diabetic mice with reduced regulatory T-cell (Treg) numbers, we found that a human FT1D-like phenotype can be induced by islet-associated autoimmunity through collaboration between innate immunity (macrophages and/or natural killer cells) and acquired immunity (predominantly cytotoxic CD8+ T cells) in genetically predisposed individuals of autoimmune type 1 diabetes with low Tregs or Treg dysfunction. To clarify greater details regarding the association of autoimmunity in the pathogenesis of FT1D, further studies using suitable animal models and accumulation of the relevant patients are required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fulminant type 1 diabetes (FT1D) is a novel subtype of type 1 diabetes and is characterized by a relatively low glycated hemoglobin (HbA1c) level at the onset, despite the abrupt occurrence of marked hyperglycemia with ketosis or ketoacidosis [1]. According to a nationwide survey of FT1D, ~ 72% of the patients with this diabetes have preceding flu-like symptoms, suggesting the involvement of viral infection in the development of the disease [2]. Moreover, 98% of the patients show increased levels of serum exocrine pancreatic enzymes, which is consistent with the reported findings of mononuclear infiltration in the exocrine pancreas [3]. Imagawa et al. initially reported that islet-associated autoantibodies were not detected and that insulitis was not observed on pancreatic biopsies within 5 months of the onset of diabetes. Thus, this type of diabetes was originally described as a subtype of type 1B diabetes [3]. In terms of pathogenesis, it was believed that viral infection and subsequent non-autoimmune inflammatory reactions in genetically susceptible individuals directly cause β-cell destruction, leading to the development of “classic” FT1D [4]. However, Tanaka et al. previously demonstrated T-cell insulitis at autopsy in a patient who died 40 min after arrival at the hospital [5], and a nationwide survey revealed that approximately 5% of patients had anti-glutamic acid decarboxylase (GAD) antibody in their serum [2], suggesting that islet-associated autoimmunity might be involved in some patients with FT1D. In the present review, we discuss a possible involvement of islet-associated autoimmunity in the development of FT1D based on the findings derived from animal models and clinical studies.
Encephalomyocarditis virus-induced diabetes murine model mimics human FT1D
The encephalomyocarditis (EMC) virus exclusively infects and destroys pancreatic β-cells, leading to the development of diabetes in genetically predisposed mice [6]. Shimada and Maruyama previously proposed the possibility that the encephalomyocarditis (EMC)-virus-induced diabetes murine model mimics human FT1D [7]. Intraperitoneal injection of male DBA/2 mice with a diabetic strain of EMC virus (EMC-D) can induce the rapid onset of diabetes a few days after administration (Fig. 1). Notably, prior to hyperglycemia onset, the blood glucose level decreased into the hypoglycemic range (Fig. 1, arrow). This may be because of temporal hyperinsulinemia attributed to the abrupt destruction of virally infected β-cells. A similar phenomenon can be observed in some patients with FT1D; i.e., hypoglycemia was observed just before disease onset [8]. A rapid onset pattern and the involvement of exocrine tissue damage, as indicated by the high serum amylase level [7], suggest that the EMC virus-induced diabetes model mimics human FT1D. Additionally, inflammatory change in the exocrine pancreas was reportedly observed in this murine model [9].

Changes in mean blood glucose level after intraperitoneal injection of EMC virus [7]. Intraperitoneal injection of male DBA/2 mice with a diabetic strain of EMC virus (EMC-D) induced rapid onset of diabetes. The arrow indicates the transient fall of blood glucose level after injection. EMC, encephalomyocarditis
Although ~ 70% of patients with FT1D have a history of antecedent flu-like symptoms [2] indicating the involvement of preceding viral infection, the etiology of the disease remains controversial. Furthermore, there is discussion on the involvement of autoimmunity as an alternative cause rather than viral infection [10]. As islet-associated autoantibody (insulin autoantibody) is detected in the serum of an EMC virus-induced diabetes murine model [11], islet-associated autoimmunity might be also associated with diabetes in this model, although viral infection induces diabetes. Hence, we would like to propose anew that EMC virus-induced diabetes model of mice is useful as one of human FT1D, and may clarify the possible involvement of autoimmunity in the development of FT1D.
Nonobese diabetic mice with reduced regulatory T-cell numbers mimic human FT1D
Several animal models of FT1D have been proposed. For instance, aggressive autoimmune diabetes and widespread destruction of α-, β-pancreatic cells and exocrine tissue can be induced by the adoptive transfer of BDC2.5 T-cell receptor-transgenic T cells, derived from an insulin granule-reactive T-cell clone, into neonate nonobese diabetic (NOD) mice [12]. These findings suggest that FT1D could be caused by the islet-associated autoimmunity in a situation of immature regulatory T cell (Treg) function.
Alternatively, we focused on CD28-deficient NOD (CD28−/− NOD) mice as a possible model of human FT1D [13]. Previous studies revealed that these mice can develop autoimmune diabetes via the development of insulitis and inflammation of the exocrine pancreas because of reduced Treg cell count [14, 15]. Due to the rapid onset of FT1D in humans and the proposed role of viral infection in disease pathogenesis, we examined the effect of administration of polyinosinic-polycytidylic acid [poly(I:C)], a mimic of double-stranded RNA, in CD28−/− NOD mice [13]. This molecule mimics a candidate virus that might be related to an RNA enterovirus that causes FT1D [16], which binds to toll-like receptor 3 expressed predominantly in macrophages or dendritic cells and activates the production of various pro-inflammatory cytokines [17]. Thus, we examined the effect of poly(I:C) treatment on the disease phenotype of CD28−/− NOD mice.
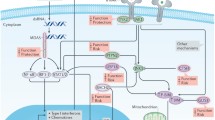
Accordingly, an injection of poly(I:C) into CD28−/− NOD mice at 8 weeks old, resulted in a rapid development of diabetes within 1–6 days, indicating a human FT1D-like phenotype. In the pancreas, CD11b (presumably macrophages) and T cells (CD4+ and CD8+ T cells) infiltrated the insulitic lesions. Moreover, DX5-positive natural killer (NK) cells, involved in innate immunity, infiltrated the exocrine and endocrine pancreas. Flow cytometry demonstrated that the population of NK cells was significantly greater in the poly(I:C)-injected group than the control group, suggesting that NK cells were likely to be involved in the pathogenesis. Moreover, mRNA expression levels of interleukin (IL)-18 and granzyme B in the pancreas were significantly higher in the poly(I:C)-injected group than the control group. IL-18, a pro-inflammatory cytokine, is produced in macrophages and dendritic cells and involved in the development of autoimmune (type 1) diabetes via islet-associated T-helper 1 (Th1) type acquired immunity [18]. Additionally, granzyme B is a caspase-like serine protease that is released by cytotoxic lymphocytes, i.e., NK and cytotoxic CD8+ T cells, to kill their target cells [19]. Thus, poly(I:C) administration might activate macrophages/dendritic cells to produce IL-18, resulting in activation and expansion of NK cells and cytotoxic CD8+ T cells in Treg-deficient conditions and causing aggressive development of diabetes mimicking human FT1D (Fig. 2). Hence, it is suggested that a FT1D-like phenotype can be caused by autoimmunity through collaboration between innate and acquired immunities in people at genetically high risk for type 1 diabetes with low Tregs or Treg dysfunction after viral infection. In fact, a significant reduction for Foxp3 expression level was reportedly observed in peripheral blood mononuclear cells of FT1D patients, suggesting the possibility that impaired Treg development/functionality might contribute to the pathogenesis of human FT1D [20]. However, a further detailed evaluation is required to examine this concept in a clinical setting.

Scheme of presumed mechanism for the development of fulminant type 1 diabetes (FT1D) via autoimmunity. Viral infection activates macrophages/dendritic cells via binding to TLR3 to produce IL-18, resulting in activation and expansion of NK cells, CD4+ T cells, and cytotoxic CD8+ T cells in Treg-deficient condition, and development of FT1D predominantly in genetically susceptible individuals of autoimmune type 1 diabetes. DC, dendritic cell; IL-18, interleukin-18; IFN, interferon; MΦ, macrophage; MHC, major histocompatibility complex; NK, natural killer; poly(I:C), polyinosinic-polycytidylic acid; Th1, T helper-1; TLR, toll-like receptor; Treg, regulatory T cell
Clinical characteristics of anti-glutamic acid decarboxylase antibody-positive FT1D
A nationwide survey performed in Japan revealed that FT1D accounts for 20% of newly diagnosed type 1 diabetes cases exhibiting hyperglycemia and diabetic ketosis/ketoacidosis [2]. Recently, as patients with FT1D increase, cases positive for islet-associated autoantibodies, predominantly anti-GAD, have been reported. Indeed, the nationwide survey of FT1D revealed that ~ 5% of FT1D patients had anti-GAD antibody in their serum [2]. Moreover, another study demonstrated that cases exhibiting anti-GAD antibody-positive FT1D accounted for 9% of FT1D cases [21]. These findings served as a basis for us to investigate the possibility that the involvement of islet-associated autoimmunity in the development of anti-GAD antibody-positive FT1D may produce some characteristic clinical signs at its onset. Hence, we investigated the relationship between anti-GAD antibody titers and clinical parameters in this type of FT1D at the onset, particularly in terms of clinical severity [22].
We determined that anti-GAD antibody titers were significantly and inversely correlated with blood pH and HCO3− levels, and significantly positively correlated with serum levels of total ketone bodies, acetoacetate, and 3-hydroxybutyrate. Moreover, this study also showed a significant inverse correlation between titers and the period from the appearance of any prodromal symptoms to diagnosis (Fig. 3). Hence, higher titers of anti-GAD antibody might be associated with more severe injury to β-cells and lower insulin secretion capability. In other words, these might further support the possible involvement of islet-associated autoimmune processes in anti-GAD antibody-positive FT1D.

Correlations between anti-GAD antibody titers and clinical parameters at diagnosis of FT1D. Scatter plots (semi-log graph) of anti-GAD antibody titers and clinical parameters were composed. a anti-GAD antibody titers vs. the period from the development of any prodromal symptoms to the diagnosis of FT1D (rs = − 0.559, p < 0.05 using the Spearman’s rank correlation test). b anti-GAD antibody titers vs. blood pH (rs = − 0.576, p < 0.05). c anti-GAD antibody titers vs. blood HCO3− (rs = − 0.578, p < 0.05). d Anti-GAD antibody titers vs. serum levels of ketone bodies. Open circle with solid line; vs. total ketone body (rs = 0.661, p < 0.05). Open square with a dotted line; vs. acetoacetate (rs = 0.700, p < 0.05). Filled triangle with a dashed line; vs. 3-hydroxybutyrate (rs = 0.782, p < 0.01). GAD, glutamic acid decarboxylase; GADA, anti-glutamic acid decarboxylase antibody; rs, Spearman’s rank correlation coefficient. Reprinted with permission from Endocr J. 2019;66(4):329–336 [22]
A previous study demonstrated that HLA-DRB1*04:05 (or DR4) and DRB1*09:01 (or DR9) are the predominant HLA alleles associated with autoimmune type 1 diabetes, whereas only DRB1*04:05 was reportedly associated with “classic” FT1D in Japan [23]. However, it was recently reported that DRB1*09:01 is strongly associated with anti-GAD antibody-positive FT1D [24]. Similarly, DRB1*09:01 was observed in 42.9% of cases in our previous study [22]. Given that GAD-reactive interferon-γ-secreting CD4+ T cells were increased in some patients with anti-GAD antibody-positive acute-onset type 1 diabetes with HLA-DR9 compared to those without it [25], GAD-associated T-cell autoimmunity may be involved in the development of anti-GAD antibody-positive FT1D in cases with HLA-DR9.
Two cases of FT1D without islet-associated autoantibodies suggest the involvement of autoimmunity
We previously encountered two cases of ‘classic’ FT1D suggesting the involvement of autoimmunity [26]. The first case showed negative islet-associated autoantibodies (anti-GAD antibody, anti-insulinoma-associated protein-2 (IA-2) antibody, islet-cell antibody, and insulin autoantibody) at the onset of the disease, although anti-GAD antibody was detected in the serum 1 year after the onset, suggesting possible autoimmunity (Fig. 4). The second case also showed negative anti-GAD, and negative anti-IA‐2 antibodies. However, high levels of serum CXCL10 and GAD‐reactive CD4+ T cells were detected in the peripheral blood. Serum level of CXCL10, an important chemokine that induces migration of activated T cells to local lesions, and GAD‐reactive CD4+ interferon‐γ‐producing cells in the peripheral blood are effective markers for T-cell‐mediated autoimmunity in type 1 diabetes [27]. Thus, FT1D should not be considered “idiopathic” at the onset, according to the absence of detectable islet‐associated autoantibodies. Hence, careful periodic measurement of islet‐associated autoantibodies and assessment of cellular immunity against islet‐associated antigens should be performed.

Changes in anti-GAD antibody titer in a patient with FT1D [26]. A 48‐year‐old man was admitted to our hospital with complaints of fever, abdominal discomfort, thirst, polyuria, and weight loss over the preceding 10 days. Based upon the presence of diabetic ketoacidosis, relatively low HbA1c level (7.3%) at the onset, absence of islet-associated autoantibodies including anti-GAD antibody, and extremely low level of serum and urinary C‐peptide levels, he was diagnosed as FT1D. Seroconversion of anti-GAD antibody was observed 1 year after the onset of the disease. Anti-GAD antibody, anti-glutamic acid decarboxylase antibody; FT1D, fulminant type 1 diabetes
Immune checkpoint inhibitor-associated FT1D
Recent studies have shown that anti-programmed cell death (PD)-1/anti-PD-ligand 1 therapy for malignancies leads to the development of FT1D [28]. We previously presented a case with anti-PD-1 (pembrolizumab) therapy-related FT1D prior to the development of diabetic ketosis or ketoacidosis, in which a detailed time course of decline in serum C-peptide levels were being examined [29]. Insulin therapy began post hyperglycemia, preventing diabetic ketoacidosis. However, the serum C-peptide levels rapidly decreased thereafter, and insulin secretion capacity was exhausted within 16 days following hyperglycemia onset. Thus, this case was considered as anti-PD-1 therapy-related FT1D. Cellular autoimmunity against islet-associated autoantigens can cause FT1D as anti-PD-1 therapy targets T-cell immunoregulatory molecules.
Conclusions
Based on the findings derived from animal models and clinical research, it appears that anti-islet-associated autoimmunity might be associated with the partial development of FT1D with or without anti-islet autoantibodies. To establish greater details regarding the pathogenesis, investigation of a suitable animal model and accumulation of the relevant cases are required.
References
Imagawa A, Hanafusa T, Awata T, Ikegami H, Uchigata Y, Osawa H, et al. Report of the committee of the japan diabetes society on the research of fulminant and acute-onset type 1 diabetes mellitus: new diagnostic criteria of fulminant type 1 diabetes mellitus (2012). J Diabetes Investig. 2012;3(6):536–9. https://doi.org/10.1111/jdi.12024.
Imagawa A, Hanafusa T, Uchigata Y, Kanatsuka A, Kawasaki E, Kobayashi T, et al. Fulminant type 1 diabetes: a nationwide survey in Japan. Diabetes Care. 2003;26(8):2345–52. https://doi.org/10.2337/diacare.26.8.2345.
Imagawa A, Hanafusa T, Miyagawa J, Matsuzawa Y. A novel subtype of type 1 diabetes mellitus characterized by a rapid onset and an absence of diabetes-related antibodies Osaka IDDM Study Group. N Engl J Med. 2000;342(5):301–7. https://doi.org/10.1056/NEJM200002033420501.
Imagawa A, Hanafusa T. Pathogenesis of fulminant type 1 diabetes. Rev Diabet Stud. 2006;3(4):169–77. https://doi.org/10.1900/RDS.2006.3.169.
Tanaka S, Kobayashi T, Momotsu T. A novel subtype of type 1 diabetes mellitus. N Engl J Med. 2000;342(24):1835–7.
Yoon JW, Jun HS. Viruses in type 1 diabetes: brief review. ILAR J. 2004;45(3):343–8. https://doi.org/10.1093/ilar.45.3.343.
Shimada A, Maruyama T. Encephalomyocarditis-virus-induced diabetes model resembles “fulminant” type 1 diabetes in humans. Diabetologia. 2004;47(10):1854–5. https://doi.org/10.1007/s00125-004-1538-9.
Hirota H, Tsutsumi C, Kimata H, Watanabe D, Imbe H, Takamto S, et al. A case of fulminant type 1 diabetes patient accompanied by hyperinsulinemic hypoglycemia prior to clinical diagnosis of diabetes. J Japan Diab Soc. 2016;59:210–7.
Matsuzaki H, Doi K, Doi C, Onodera T, Mitsuoka T. Susceptibility of four species of small rodents to encephalomyocarditis (EMC) virus infection. Jikken Dobutsu. 1989;38(4):357–61. https://doi.org/10.1538/expanim1978.38.4_357.
Shimada A, Morimoto J, Kodama K, Oikawa Y, Irie J, Nakagawa Y, et al. T-cell-mediated autoimmunity may be involved in fulminant type 1 diabetes. Diabetes Care. 2002;25(3):635–6. https://doi.org/10.2337/diacare.25.3.635.
Maruyama T, Takei I, Asaba Y, Yanagawa T, Takahashi T, Itoh H, et al. Insulin autoantibodies in mouse models of insulin-dependent diabetes. Diabetes Res. 1989;11(2):61–5.
Bergman B, McManaman JL, Haskins K. Biochemical characterization of a beta cell membrane fraction antigenic for autoreactive T cell clones. J Autoimmun. 2000;14(4):343–51. https://doi.org/10.1006/jaut.2000.0377.
Tada A, Shimada A, Yamada T, Oikawa Y, Yamada Y, Okubo Y, et al. A mimic of viral double-stranded RNA triggers fulminant type 1 diabetes-like syndrome in regulatory T cell-deficient autoimmune diabetic mouse. J Immunol. 2011;187(10):4947–53. https://doi.org/10.4049/jimmunol.1000837.
Meagher C, Tang Q, Fife BT, Bour-Jordan H, Wu J, Pardoux C, et al. Spontaneous development of a pancreatic exocrine disease in CD28-deficient NOD mice. J Immunol. 2008;180(12):7793–803. https://doi.org/10.4049/jimmunol.180.12.7793.
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12(4):431–40. https://doi.org/10.1016/s1074-7613(00)80195-8.
Imagawa A, Hanafusa T, Makino H, Miyagawa JI, Juto P. High titres of IgA antibodies to enterovirus in fulminant type-1 diabetes. Diabetologia. 2005;48(2):290–3. https://doi.org/10.1007/s00125-004-1624-z.
Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13(5):816–25. https://doi.org/10.1038/sj.cdd.4401850.
Oikawa Y, Shimada A, Kasuga A, Morimoto J, Osaki T, Tahara H, et al. Systemic administration of IL-18 promotes diabetes development in young nonobese diabetic mice. J Immunol. 2003;171(11):5865–75. https://doi.org/10.4049/jimmunol.171.11.5865.
Trapani JA, Sutton VR. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol. 2003;15(5):533–43. https://doi.org/10.1016/s0952-7915(03)00107-9.
Wang Z, Zheng Y, Hou C, Yang L, Li X, Lin J, et al. DNA methylation impairs TLR9 induced Foxp3 expression by attenuating IRF-7 binding activity in fulminant type 1 diabetes. J Autoimmun. 2013;41:50–9. https://doi.org/10.1016/j.jaut.2013.01.009.
Kawasaki E, Nakamura K, Kuriya G, Satoh T, Kobayashi M, Kuwahara H, et al. Differences in the humoral autoreactivity to zinc transporter 8 between childhood- and adult-onset type 1 diabetes in Japanese patients. Clin Immunol. 2011;138(2):146–53. https://doi.org/10.1016/j.clim.2010.10.007.
Saito D, Oikawa Y, Mizutani G, Inoue K, Hatano M, Inoue I, et al. Clinical characteristics of anti-glutamic acid decarboxylase antibody-positive fulminant type 1 diabetes. Endocr J. 2019;66(4):329–36. https://doi.org/10.1507/endocrj.EJ18-0417.
Kawabata Y, Ikegami H, Awata T, Imagawa A, Maruyama T, Kawasaki E, et al. Differential association of HLA with three subtypes of type 1 diabetes: fulminant, slowly progressive and acute-onset. Diabetologia. 2009;52(12):2513–21. https://doi.org/10.1007/s00125-009-1539-9.
Tsutsumi C, Imagawa A, Ikegami H, Makino H, Kobayashi T, Hanafusa T, et al. Class II HLA genotype in fulminant type 1 diabetes: a nationwide survey with reference to glutamic acid decarboxylase antibodies. J Diabetes Investig. 2012;3(1):62–9. https://doi.org/10.1111/j.2040-1124.2011.00139.x.
Itoh A, Shimada A, Kodama K, Morimoto J, Suzuki R, Oikawa Y, et al. GAD-reactive T cells were mainly detected in autoimmune-related type 1 diabetic patients with HLA DR9. Ann N Y Acad Sci. 2004;1037:33–40. https://doi.org/10.1196/annals.1337.006.
Nakagawa Y, Shimada A, Oikawa Y, Irie J, Shigihara T, Tsumura K, et al. Two cases of “fulminant” type 1 diabetes suggesting involvement of autoimmunity. Ann N Y Acad Sci. 2003;1005:359–61. https://doi.org/10.1196/annals.1288.059.
Shimada A, Morimoto J, Kodama K, Suzuki R, Oikawa Y, Funae O, et al. Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care. 2001;24(3):510–5. https://doi.org/10.2337/diacare.24.3.510.
Baden MY, Imagawa A, Abiru N, Awata T, Ikegami H, Uchigata Y, et al. Characteristics and clinical course of type 1 diabetes mellitus related to anti-programmed cell death-1 therapy. Diabetol Int. 2019;10(1):58–66. https://doi.org/10.1007/s13340-018-0362-2.
Saito D, Oikawa Y, Yano Y, Ikegami Y, Satomura A, Isshiki M, et al. Detailed time course of decline in serum c-peptide levels in anti-programmed cell death-1 therapy-induced fulminant type 1 diabetes. Diabetes Care. 2019;42(3):e40–e4141. https://doi.org/10.2337/dc18-1673.
Funding
No specific funding or Grant was received for this work.
Author information
Authors and Affiliations
Contributions
YO wrote the manuscript. AS reviewed the manuscript and contributed to discussions.
Corresponding author
Ethics declarations
Conflict of interest
A.S. has received lecture fees from Astellas Pharm Inc., Eli Lilly Japan K.K., Ono pharmaceutical Co., Ltd., Terumo Corporation and Sanofi K.K. A.S. has received research funding from Astellas Pharm inc. and Mitsubishi Tanabe Pharma Corporation. A.S. has received scholarship grants from Astellas Pharm inc., Daiichi Sankyo Co., Ltd. and Kyowa Kirin Co., Ltd., MSD K.K., Novo Nordisk Pharma Ltd. and Ono Pharmaceutical Co., LTD. Y.O. declares no conflicts of interest relevant to this article.
Ethics policy
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Oikawa, Y., Shimada, A. Possible involvement of autoimmunity in fulminant type 1 diabetes. Diabetol Int 11, 329–335 (2020). https://doi.org/10.1007/s13340-020-00460-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13340-020-00460-8