
Abstract
Introduction
This multicenter, randomized study assessed the efficacy and safety of the dipeptidyl peptidase 4 inhibitor sitagliptin added to insulin monotherapy in Japanese patients with type 2 diabetes mellitus (T2DM).
Materials and methods
This study had an initial 16-week, double-blind treatment period in which 266 patients on diet/exercise and insulin monotherapy for ≥12 weeks were randomized (1:1) to sitagliptin 50 mg q.d. (N = 129; mean baseline HbA1c = 8.9 %) or placebo (N = 137; mean baseline HbA1c = 8.9 %). It was followed by a 36-week, open-label treatment period in which all patients received sitagliptin 50 mg q.d., which could have been increased to 100 mg q.d. for patients meeting predefined glycemic criteria.
Results
After 16 weeks, treatment with sitagliptin resulted in significant placebo-adjusted mean decreases from baseline in HbA1c, fasting plasma glucose, and 2-h postmeal glucose of −0.9 % (p < 0.001), −11.4 mg/dl (p = 0.007), and −39.9 mg/dl (p < 0.001), respectively. During the double-blind period, adverse experiences (AEs) were reported with similar frequency in both treatment groups and the incidence of gastrointestinal AEs was low. The incidence of hypoglycemia AEs in the sitagliptin group (20.2 %) was higher than in the placebo group (12.4 %), but the between-group difference was not statistically significant (p = 0.097). Small increases from baseline in body weight were observed with sitagliptin [sitagliptin: 0.6 kg (p < 0.001), placebo: 0.1 kg (p = 0.498)]. In the open-label period, sustained improvements in glycemic parameters were observed with sitagliptin treatment, and sitagliptin was generally well tolerated.
Conclusions
In Japanese patients with T2DM inadequately controlled on insulin monotherapy, the addition of sitagliptin provided significant improvements in glycemic parameters and was generally well tolerated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insulin therapy is widely used in Japan for the treatment of patients with type 2 diabetes mellitus (T2DM). Of Japanese patients with T2DM receiving pharmacological treatment, 31 % are treated with insulin as monotherapy or combination therapy with oral antihyperglycemic agents (OHAs) [1, 2]. Treatment regimens with basal or pre-mixed insulin effectively lower fasting glucose levels. Although pre-mixed insulin may have some effect in lowering post-meal glucose, it may not adequately control glycemic excursions in the postprandial state. Insulin therapy in combination with OHAs is effective, but many patients still fail to reach currently recommended HbA1c treatment goals.
The incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), which are released by the intestine in response to a meal, play an important role in glycemic homeostasis through multiple physiological pathways, including stimulation of insulin secretion (GLP-1 and GIP) and suppression of glucagon secretion (GLP-1) [3–5]. These beneficial actions are limited by the rapid degradation of intact GLP-1 and GIP by the peptidase enzyme, dipeptidyl peptidase-4 (DPP-4) [3]. DPP-4 inhibitors are a novel therapeutic class of drugs for the treatment of T2DM that act to stabilize the intact forms of GLP-1 and GIP and thereby improve glycemic control [6, 7].
Sitagliptin is an oral, highly selective DPP-4 inhibitor for the treatment of T2DM. Several large, randomized, placebo-controlled trials have demonstrated that treatment with sitagliptin as monotherapy or in combination with other OHAs is generally well tolerated and provides significant improvement in key glycemic parameters compared with placebo [8–20].
The efficacy and safety of sitagliptin added to ongoing insulin therapy with or without concomitant metformin have been demonstrated in non-Japanese T2DM patients [20]. The efficacy and safety of sitagliptin or other DPP-4 inhibitors added to insulin monotherapy have not been studied in Japanese T2DM patients. The glucose-lowering actions of sitagliptin and exogenously administered insulin may be complementary. In addition, the pathological mechanisms underlying T2DM may differ in Japanese patients relative to those from other ethnic and genetic backgrounds [21–24]. The present study examined the efficacy and safety of the addition of sitagliptin to Japanese patients with T2DM who were inadequately controlled on insulin monotherapy.
Materials and methods
Patients
This study enrolled Japanese patients ≥20 years of age with T2DM [HbA1c ≥7.9 to <10.5 % and fasting plasma glucose (FPG) ≥126 to ≤220 mg/dl] who were on both diet/exercise and taking insulin monotherapy [pre-mixed (containing 25 or 30 % of fast-acting or ultra-short-acting insulin), intermediate-acting, or long-acting type insulin] for ≥12 weeks. This study used Japan Diabetes Society (JDS)-certified HbA1c values, the standard at the time the study was conducted. HbA1c values reported here have been converted to National Glycohemoglobin Standardization Program (NGSP) values as follows: HbA1c [NGSP] [%] = 1.02 × HbA1c [JDS] [%] + 0.25 % [25]. Main exclusion criteria included history of type 1 diabetes; treatment with fast-acting, ultra-short-acting, or pre-mixed type insulin that did not contain 25 % or 30 % of fast-acting or ultra-short-acting insulin within 12 weeks before the start of the treatment period; presence of progressive diabetes complications; unstable cardiovascular disease or uncontrolled severe hypertension; increased serum creatinine (>1.5 mg/dl in men or >1.3 mg/dl in women) or increased alanine aminotransferase or aspartate aminotransferase >twofold the upper limit of normal; hemoglobin <11.0 g/dl in men or <10.0 g/dl in women; or body mass index (BMI) <18 or >40 kg/m2.
Study design and procedures
This multicenter, randomized clinical trial (registered at http://www.clinicaltrials.gov as NCT00854035) was conducted at 60 sites in Japan.
The overall study design is shown in Fig. 1. Patients who met all eligibility criteria entered a 2-week, single-blind, placebo run-in period. Otherwise, patients on combination therapy with insulin and other OHAs, with HbA1c values ≥7.4 and ≤9.4 % and who met all other eligibility criteria, could enter the placebo run-in period following a 10-week wash-out period of non-insulin OHAs. This design ensured that all patients received at least 12 weeks of diet/exercise therapy and at least 12 weeks of insulin therapy at a stable dose prior to randomization. All patients were instructed to follow a stable program of diet and exercise for the duration of the study.

Study design
Patients were eligible for randomization if they had an HbA1c ≥7.9 and <10.5 % and a FPG ≥126 and ≤220 mg/dl just prior to initiating the placebo run-in and ≥75 % treatment compliance (based on pill counts) during the placebo run-in. Eligible patients were randomized (1:1) to either sitagliptin 50 mg q.d. or matching placebo for 16 weeks in double-blind fashion using a computer-generated allocation schedule.
Upon completion of the double-blind period, patients entered a 36-week, open-label treatment period. Patients who received sitagliptin during the double-blind period continued to do so in the open-label period (S/S group). Patients who received placebo in the double-blind period were started on sitagliptin 50 mg q.d. upon entry to the open-label period (P/S group). The dose of sitagliptin in the open-label period was up-titrated from 50 to 100 mg for patients meeting protocol-specified criteria: FPG ≥140 mg/dl from week 20 through week 32 or HbA1c ≥7.4 % from week 28 through week 32. The insulin dose was to remain stable throughout the study, except if the insulin dose needed to be reduced because of the occurrence of, or for prevention of, hypoglycemia. Patients not meeting progressively stricter glycemic goals had an up-adjustment to their insulin dose (glycemic rescue), based on the clinical judgment of the investigator and the following FPG criteria: FPG >240 mg/dl two consecutive times from randomization (day 1) through week 24 or FPG >200 mg/dl two consecutive times from week 24 through to the end of study. If up-titration criteria for insulin and for the study drug were met simultaneously, the dose of insulin as rescue therapy should have been increased prior to considering increasing the dose of study drug.
Meal tolerance tests were performed at weeks 0, 16, and 52 or at the visit for discontinuation, starting 30 min after administration of study drug (at week 0 patients received a dose of matching placebo). The test meal contained ~500 kcal (60 % carbohydrate, 15 % protein, and 25 % fat) and was to be consumed within 15 min. Blood samples were drawn prior to beginning the test meal and 0.5, 1, and 2 h after beginning the meal.
The study was conducted in accordance with principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies. All patients provided written informed consent.
Study endpoints
Change from baseline in HbA1c at week 16 was the primary efficacy endpoint, and changes from baseline in FPG and 2-h postmeal glucose (2-h PMG) at week 16 were secondary endpoints. In the open-label period HbA1c, FPG, and 2-h PMG were assessed as exploratory endpoints. Additionally, fasting 1,5-anhydroglucitol, total 2-h postmeal glucose AUC, and total 2-h postmeal C-peptide AUC were assessed as exploratory endpoints at weeks 16 and 52. The proportions of patients with HbA1c values meeting the therapeutic goals of <7.4 and <6.9 %, [corresponding to 7.0 and 6.5 % in HbA1c (JDS), respectively] also were assessed at week 16 and week 52.
Adverse experiences (AEs) were monitored throughout the study up to 2 weeks post-treatment and were rated by investigators as to their intensity and relationship to study drug. Hypoglycemia and selected gastrointestinal AEs (nausea, vomiting, and diarrhea) were predefined as adverse events of interest. Hypoglycemia was diagnosed by the investigators based on their assessment of patients’ reports. Patients were instructed to notify the investigator immediately if they had symptoms consistent with hypoglycemia (e.g., sweating, anxiety, palpitations, headache, blurred vision, loss of consciousness) and had self-monitoring blood glucose values <70 mg/dl or if they had self-monitoring blood glucose values >240 mg/dl (from weeks 0 to 24) or >200 mg/dl (from weeks 24 to 52) two consecutive times. During the study, the safety and tolerability were also assessed by physical examination, monitoring of vital signs, ECG, and safety laboratory tests that included hematology, serum chemistry, and urinalysis.
All laboratory assays were performed at one central laboratory (Mitsubishi Chemical Medience Corp., Tokyo, Japan).
Statistical methods
Efficacy
The efficacy analysis included all randomized patients who had taken at least one dose of study drug and had a baseline measurement or at least one measurement post-randomization. A constrained longitudinal data analysis (cLDA) method [26] with terms for treatment, time, and the interaction of time by treatment was used to evaluate continuous outcome change from baseline at week 16. The analysis model also adjusted for prior OHAs status (yes/no) and type of insulin. Missing values were handled by the cLDA model, without explicit imputation. The between-group difference in least squares (LS) means and the corresponding 95 % confidence interval (CI) was estimated from the cLDA model. A p value <0.05 (two-sided) was considered statistically significant. The proportions of patients with HbA1c values meeting the HbA1c goals of <7.4 and <6.9 % at week 16 were analyzed using a logistic regression model that included treatment group, prior oral OHA status, type of insulin, and baseline HbA1c as covariates. For this analysis, measurements at both baseline and week 16 were needed. For HbA1c, FPG, and 2-h PMG, the subgroup analysis of insulin type (i.e. premixed or non-premixed) was assessed by using the same cLDA method described above for each subgroup. The subgroup analyses for FPG and 2-h PMG were post hoc.
For long-term efficacy assessment, summary statistics for efficacy endpoints were provided by treatment group (P/S or S/S) at each time point in which the endpoint was measured up to week 52; missing values were not imputed. At week 52, the within-group mean change from baseline (i.e., week 0) for all efficacy endpoints was assessed using a paired-Student’s t test. For P/S, comparisons versus baseline (week 0) were performed post hoc.
The effect of increasing the dose of sitagliptin to 100 mg q.d. was assessed (post hoc). Among those patients whose sitagliptin dose was increased and whose HbA1c value at the time of up-titration was ≥7.4 %, the proportion of patients with HbA1c values <7.4 % at 16 weeks after up-titration was tabulated. Additionally, for patients whose sitagliptin dose was increased and who completed the study, the proportion of patients with HbA1c values <7.4 % at week 52 was also assessed. For these analyses, missing values were not imputed.
Post-glycemic rescue data were excluded for all evaluations of efficacy parameters.
Safety
For the double-blind period, safety and tolerability analyses included all randomized patients. Between-group comparisons using Fisher’s exact test were performed for the percentages of patients with one or more AEs, drug-related AEs, AEs of hypoglycemia, and prespecified gastrointestinal AEs. For laboratory tests, vital signs, and body weight, summary statistics were generated up to week 16 for each group individually.
For the long-term safety assessment, the patient population included all patients who received at least one dose of sitagliptin in the open-label portion of the study (i.e., from week 16 to week 52). Safety data were summarized by treatment group.
Post-glycemic rescue data were excluded for evaluations of hypoglycemia and body weight.
Results
Five hundred thirty-two patients were screened, of whom 266 were randomized to treatment (129 to sitagliptin and 137 to placebo) (Fig. 2). Demographic, anthropometric, and disease characteristics were generally similar between the two treatment groups (Table 1). Patients had mild to moderate hyperglycemia with a baseline mean HbA1c of 8.9 % and mean FPG of 164.9 mg/dl. The average duration of known diabetes was 14.0 years and the mean BMI was 25.2 kg/m2.

Patient disposition. Patients in the P/S group received placebo during the double-blind period and sitagliptin in the open-label period. Patients in the S/S group received sitagliptin in both periods
Two hundred fifty-four patients completed the double-blind period and entered the open-label period; of those, 239 patients subsequently completed the open-label period (Fig. 2).
Efficacy
Double-blind period (weeks 0 through 16)
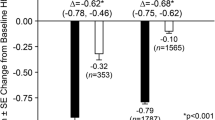
The addition of sitagliptin to Japanese patients receiving insulin therapy resulted in a significant (p < 0.001) reduction from baseline in HbA1c compared with placebo at week 16 (Table 2; Fig. 3). The between-group difference in LS mean (95 % CI) change from baseline in HbA1c at week 16 was −0.9 % (−1.0, −0.7). At week 16, a significantly greater proportion of patients in the sitagliptin 50 mg group relative to the placebo group had HbA1c values meeting the goals of <7.4 % (24.0 vs. 2.3 %, respectively; p < 0.001), and <6.9 % (8.0 vs. 1.6 %, respectively; p = 0.013).

Time course of HbA1c in Japanese patients with type 2 diabetes mellitus treated with double-blind sitagliptin 50 mg q.d. or placebo added to insulin for the first 16 weeks and open-label sitagliptin 50 or 100 mg q.d. added to insulin for the subsequent 36 weeks. The data are values for mean ± SE and results from the S/S and P/S treatment groups are indicated with open triangles and closed circles, respectively. The data that were obtained after initiation of additional antihyperglycemic agents are excluded
Significant improvements in both FPG and 2-h PMG were also observed with sitagliptin treatment at week 16 relative to placebo (p < 0.01 for FPG and p < 0.001 for 2-h PMG), with between-group differences in LS-mean (95 % CI) changes from baseline of −11.4 mg/dl (−19.7, −3.1) and −39.9 mg/dl (−52.6, −27.2), respectively (Table 2; Figs. 4, 5).

Time course of fasting plasma glucose (FPG) results in Japanese patients with type 2 diabetes mellitus treated with double-blind sitagliptin 50 mg q.d. or placebo added to insulin for the first 16 weeks and open-label sitagliptin 50 or 100 mg q.d. added to insulin for the subsequent 36 weeks. The data are values for mean ± SE. Results from the S/S and P/S treatment groups are indicated with open triangles and closed circles, respectively. The data that were obtained after initiation of additional antihyperglycemic agents are excluded

Time course of meal tolerance results in Japanese patients with type 2 diabetes mellitus treated with double-blind sitagliptin 50 mg q.d. or placebo added to insulin for the first 16 weeks and open-label sitagliptin 50 or 100 mg q.d. added to insulin for the subsequent 36 weeks. The data are values for mean ± SE
The changes from baseline and between-group differences for HbA1c, FPG, and 2-h PMG at week 16 in the subgroups of patients on premixed or non-premixed insulin (Table 3) were consistent with those seen in the entire cohort (Table 2).
Consistent results were observed in analyses of changes from baseline in other efficacy parameters between the treatment groups at week 16 that were supportive of the primary and secondary findings. This included significant improvement with sitagliptin treatment in 1,5-anhydroglucitol, a parameter reflective of post-meal plasma glucose excursions and urinary glucose excretion, as well as significant improvements in post-meal total glucose and C-peptide AUCs (Table 2).
Open-label period (weeks 16 through 52)
Efficacy measurements in the open-label period, including HbA1c, FPG, and 2-h PMG, showed improvements from baseline to week 52 in the S/S and P/S groups (p < 0.001 for HbA1c and 2-h PMG, and p < 0.01 for FPG, Table 4; Figs. 3, 4, 5). Similarly, in both the S/S and P/S groups at week 52, significant changes (p < 0.05) from baseline were observed in other efficacy parameters (i.e., 1,5-anhydroglucitol and post-meal total glucose AUC; Table 4). Changes from baseline to week 52 in C-peptide AUC were significant in the S/S group (p < 0.001) but not significant in the P/S group (p = 0.163) (Table 4). Mean HbA1c decreased to similar levels in both the S/S group and the P/S groups by week 28, and remained similar thereafter (Fig. 3). However, this result should be viewed with caution since, at week 16, the groups were not necessarily representative of randomized populations and the timing of initiation of sitagliptin treatment was different in both groups.
In order to provide additional glycemic efficacy, dose titration of sitagliptin to 100 mg was allowed after week 20 for patients meeting predefined criteria of glycemic parameters. The sitagliptin dose was up-titrated in 113 patients in the S/S group and 113 patients in the P/S group. HbA1c values at 16 weeks post-escalation were obtained from a total of 205 patients (S/S group: 105 patients, P/S group: 100 patients). Overall, 11.6 % (20/172) of patients with an HbA1c ≥7.4 % before up-titration achieved HbA1c <7.4 % (treatment target achievement rate) 16 weeks after up-titration. Additionally, among all patients whose dose was up-titrated, 198 patients completed the study; 21.2 % (42/198) patients had an HbA1c <7.4 % at week 52.
Safety
Double-blind period (weeks 0 through 16)
In the double-blind period (week 0 to week 16), clinical AEs overall were reported for 58.9 % (76/129) in the sitagliptin group and 51.8 % (71/137) in the placebo group (Table 5). The incidences of drug-related clinical AEs were 16.3 % (21/129) in the sitagliptin group and 11.7 % (16/137) in the placebo group (Table 5). There were no statistically significant between-group differences in the incidences of overall clinical or drug-related AEs at week 16 (overall clinical AEs, p = 0.268; overall drug-related clinical AEs, p = 0.293).
In the double-blind period, the incidences of serious clinical AEs were similar in both treatment groups [3.1 % (4/129) in the sitagliptin group and 2.2 % (3/137) in the placebo group, Table 5]. No serious drug-related clinical AEs were reported for patients in the sitagliptin group. Three patients discontinued because of a clinical AE, one (malignant neoplasm in the lung; judged unrelated to the study drug by the investigator) in the sitagliptin group and two (eczema, hypoglycemia) in the placebo group (Table 5). No patients discontinued because of a serious drug-related clinical AE (Table 5).
The incidence of hypoglycemia in the double-blind period was 26/129 (20.2 %) in the sitagliptin group and 17/137 (12.4 %) in the placebo group (Table 5); the difference between treatments was not significantly different (p = 0.097). One episode of severe hypoglycemia was reported for one patient in the sitagliptin group and one in the placebo group. All other episodes of hypoglycemia in the sitagliptin group were generally mild to moderate in intensity, and none led to discontinuation of therapy. One of the drug-related adverse experiences of hypoglycemia reported in the sitagliptin 50 mg group was severe, but resolved while the patient was on treatment and did not lead to discontinuation. The severe hypoglycemia event reported in the sitagliptin 50 mg group occurred in a 73-year-old female patient 36 days after initiation of treatment. This event was associated with hunger, sleepiness, sweating and difficulty in concentrating and walking. Self-monitored blood glucose level was 53 mg/dl. The episode lasted about 10 min, resolved without medical treatment, and did not lead to study discontinuation. This episode was considered severe because the patient required assistance from others to manage symptoms. The severe hypoglycemia event reported in the placebo group occurred in a 59-year-old male patient 23 days after initiation of treatment and lasted for 2 h. The patient was discontinued, but the AE was assessed as not related to treatment.
The incidence of prespecified gastrointestinal AEs (nausea, vomiting, and diarrhea) occurring in the double-blind period was low in both treatment groups (Table 5); these AEs were mild to moderate in intensity and did not lead to discontinuation.
During the double-blind period, the incidence of laboratory AEs was 3.9 and 3.6 % in the sitagliptin and placebo groups, respectively (Table 5). The incidence of drug-related laboratory AEs was 2.3 % in the sitagliptin group and 0.7 % in the placebo group (Table 5). No specific laboratory AE occurred in two or more patients in the sitagliptin group.
Small increases from baseline in body weight were observed with sitagliptin [sitagliptin: 0.6 kg (p < 0.001), placebo: 0.1 kg (p = 0.498)] at week 16. The magnitude of these changes was too small to be considered clinically relevant.
Open-label period (weeks 16 through 52)
Consistent with the longer period of observation in a patient population with T2DM, one or more clinical AEs were reported for most patients in both the S/S and P/S groups during the open-label period [week 16 to week 52; 87.2 % (109/125) of patients in the S/S group and 81.4 % (105/129) of patients in the P/S group; Table 5]. Clinical AEs reported with an incidence ≥5 % in either the S/S or P/S group included diabetic nephropathy, constipation, nasopharyngitis, hypoglycemia, back pain, and upper respiratory tract infection (Table 5). Drug-related clinical AEs were reported in 28.8 and 21.7 % of patients in the S/S and P/S groups, respectively (Table 5). There were no noteworthy differences in the nature of overall clinical AEs and drug-related clinical AEs between weeks 16 and 52.
Serious clinical AEs were reported for seven patients in the S/S group and nine patients in the P/S group (Table 5); no specific SAE was reported to occur in more than one patient. There was one death in the open-label period (S/S group; Table 5). The patient, a 67-year-old male with long-lasting diabetes (17.2 years) and retinopathy, nephropathy, and neuropathy due to diabetes, was found dead 13 days after the start of the open-label period. A postmortem examination indicated myocardial infarction as a probable cause of death. The investigator considered the event as possibly related to the study drug.
In the open-label period, the incidence of hypoglycemia was 26.4 % (33/125) in the S/S group and 20.9 % (27/129) in the P/S group (Table 5). All episodes of hypoglycemia in the open-label period were mild to moderate in intensity, and none led to dose reduction or discontinuation from the study.
The incidence of prespecified gastrointestinal AEs (nausea, vomiting, and diarrhea) occurring in the open label period was low in the S/S and P/S groups (Table 5).
In the open-label period, laboratory AEs were reported in 4/124 (3.2 %) patients in the S/S group and 8/129 (6.2 %) patients in the P/S group; none was serious, and only one in each group led to discontinuation (Table 5).
In the open-label period, small increases from baseline in body weight were observed in the S/S group (0.4 kg, p = 0.005) and P/S group (0.8 kg, p < 0.001) at week 52.
Discussion
The addition of sitagliptin for 16 weeks provided significant reductions in HbA1c, FPG, and 2-h PMG relative to placebo in Japanese patients with T2DM whose glycemic control was not adequate with insulin monotherapy, demonstrating that sitagliptin improves fasting and postprandial glycemic control in combination with insulin. The proportions of patients achieving the goals of HbA1c <7.4 % and <6.9 % with sitagliptin treatment were greater compared with those for placebo.
The efficacy of sitagliptin as demonstrated by changes in HbA1c, FPG, and 2-h PMG remained stable through the 52-week study period. Patients randomized in this trial were on long-acting, intermediate-acting, or premixed insulin therapy. Overall, the HbA1c, FPG, and 2-h PMG treatment responses were similar in patients treated with long-acting or intermediate-acting insulin therapies compared to those treated with premixed insulin therapy. The present findings are clinically meaningful, but should be interpreted with caution, considering the current study design, in which the dose regimen of sitagliptin (50 mg q.d.) could have been up-titrated to 100 mg q.d. in patients meeting predefined criteria of glycemic parameters. The improvement in glycemic parameters with sitagliptin for up to 52 weeks in Japanese patients with T2DM and inadequate glycemic control with insulin monotherapy is consistent with results from prior studies of sitagliptin as monotherapy [8, 11, 12, 14–16] and as add-on to metformin [9, 17], pioglitazone [13, 18], and glimepiride [10, 19] in both Japanese and non-Japanese patients.
The addition of sitagliptin to insulin monotherapy was generally well tolerated during the double-blind and open label study periods. There were no notable differences in the nature of clinical AEs or drug-related clinical AEs during the long-term administration of sitagliptin (week 52) relative to short-term use of the drug (week 16). The incidence of hypoglycemia was higher in the sitagliptin group (20.2 %) than in the placebo group (12.4 %) at week 16. However, the number of patients who had episodes of marked severity was small (one each in the sitagliptin and placebo groups), and no episodes of hypoglycemia led to discontinuation in the sitagliptin group. Similar findings were reported with sitagliptin added to a sulphonylurea, another agent associated with hypoglycemia [10, 19]. In contrast, treatment with sitagliptin as monotherapy or in combination with agents not associated with hypoglycemia (e.g., metformin and pioglitazone) had an incidence of hypoglycemia similar to that of placebo [7, 12–14], consistent with the glucose dependency of the increase in insulin secretion and the suppression of glucagon concentrations with incretin-based therapies [27]. In light of the fact that exogenously administered insulin is not regulated by ambient glucose or incretins, and given that sitagliptin improved ambient glucose concentration (fasting and postprandial states), the finding of a higher incidence of hypoglycemia with the addition of sitagliptin to ongoing insulin therapy in the present study was not unexpected. In the open-label period, there was no notable increase in the incidence of hypoglycemia with long-term administration of sitagliptin. In order to prevent hypoglycemia in clinical practice when sitagliptin is added on to ongoing insulin therapy, down-titration of insulin should be considered. The incidence of prespecified gastrointestinal AEs was low and similar between the two treatment groups. An undesired side effect of certain antihyperglycemic agents is increased body weight [28]. Weight gain is typically observed with insulin therapy because of improved glycemic control [29]. In the present study, the improved glycemic control with sitagliptin when added to insulin therapy was not associated with a clinically meaningful change in body weight relative to baseline or the placebo group.
In Japanese patients with T2DM inadequately controlled with diet and exercise and ongoing insulin therapy, the addition of sitagliptin 50 mg q.d. after 16 weeks of treatment resulted in significant reductions from baseline in HbA1c, FPG, and 2-h PMG relative to placebo, consistent with previously reported improvements in active GLP-1 with sitagliptin therapy. These improvements remained stable throughout 52 weeks of treatment. The addition of sitagliptin to insulin was generally well tolerated, with a low incidence of severe hypoglycemia and gastrointestinal AEs and no meaningful change in body weight.
References
Kanatsuka A, Kawai K, Hirao K. Research on antihyperglycemic therapies in patients with type 2 diabetes mellitus in Japan (I): drug therapies and actual drug use. J Japan Diabetes Soc. 2006;49:409–15.
Kanatsuka A, Kawai K, Hirao K. Research on antihyperglycemic drug therapies in patients with type 2 diabetes mellitus in Japan (II): the effectiveness on glycemic control (JDDM7). J Japan Diabetes Soc. 2006;49:919–27.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–57.
Herman GA, Bergman A, Stevens C, Kotey P, Yi B, Zhao P, et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:4612–9.
Herman GA, Bergman A, Liu F, Stevens C, Wang AQ, Zeng W, et al. Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects. J Clin Pharmacol. 2006;46:876–86.
Herman GA, Stein PP, Thornberry NA, Wagner JA. Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: focus on sitagliptin. Clin Pharmacol Ther. 2007;81:761–7.
Karasik A, Aschner P, Katzeff H, Davies MJ, Stein PP. Sitagliptin, a DPP-4 inhibitor for the treatment of patients with type 2 diabetes: a review of recent clinical trials. Curr Med Res Opin. 2008;24:489–96.
Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006;29:2632–7.
Charbonnel B, Karasik A, Liu J, Wu M, Meininger G. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care. 2006;29:2638–43.
Hermansen K, Kipnes M, Luo E, Fanurik D, Khatami H, Stein P. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor, sitagliptin, in patients with type 2 diabetes mellitus inadequately controlled on glimepiride alone or on glimepiride and metformin. Diabetes Obes Metab. 2007;9:733–45.
Iwamoto Y, Tajima N, Kadowaki T, Nonaka K, Taniguchi T, Nishii M, Arjona Ferreira JC, Amatruda JM. Efficacy and safety of sitagliptin monotherapy compared with voglibose in Japanese patients with type 2 diabetes: a randomized, double-blind trial. Diabetes Obes Metab. 2010;12:613–22.
Iwamoto Y, Taniguchi T, Nonaka K, Okamoto T, Okuyama K, Arjona Ferreira JC, Amatruda J. Dose-ranging efficacy of sitagliptin, a dipeptidyl peptidase-4 inhibitor, in Japanese patients with type 2 diabetes mellitus. Endocr J. 2010;57:383–94.
Kashiwagi A, Kadowaki T, Tajima N, Nonaka K, Taniguchi T, Nishii M, Arjona Ferreira JC, Amatruda JM. Sitagliptin added to treatment with ongoing pioglitazone for up to 52 weeks improves glycemic control in Japanese patients with type 2 diabetes. J Diabetes Invest. 2011;2:381–90.
Nonaka K, Kakikawa T, Sato A, Okuyama K, Fujimoto G, Kato N, et al. Efficacy and safety of sitagliptin monotherapy in Japanese patients with type 2 diabetes. Diabetes Res Clin Pract. 2008;79:291–8.
Odawara M, Kadowaki T, Tajima N, Nishii M, Taniguchi T, Arjona Ferreira JC. Long-term safety, tolerability, and efficacy of the dipeptidyl peptidase-4 inhibitor sitagliptin in Japanese patients with type 2 diabetes. Diabetol Int. 2011;2:94–105.
Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia. 2006;49:2564–71.
Raz I, Chen Y, Wu M, Hussain S, Kaufman KD, Amatruda JM, Langdon RB, Stein PP, Alba M. Efficacy and safety of sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes. Curr Med Res Opin. 2008;24:537–50.
Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther. 2006;28:1556–68.
Tajima N, Kadowaki T, Odawara M, Nishii M, Taniguchi T, Arjona Ferreira JC. Addition of sitagliptin to ongoing glimepiride therapy in Japanese patients with type 2 diabetes over 52 weeks leads to improved glycemic control. Diabetol Int. 2011;2:32–44.
Vilsboll T, Rosenstock J, Yki-Jarvinen H, Cefalu WT, Chen Y, Luo E, et al. Efficacy and safety of sitagliptin when added to insulin therapy in patients with type 2 diabetes. Diabetes Obes Metab. 2010;12:167–77.
Chen KW, Boyko EJ, Bergstrom RW, Leonetti DL, Newell-Morris L, Wahl PW, Fujimoto WY. Earlier appearance of impaired insulin secretion than of visceral adiposity in the pathogenesis of NIDDM. 5-Year follow-up of initially nondiabetic Japanese-American men. Diabetes Care. 1995;18:747–53.
Kim DJ, Lee MS, Kim KW, Lee MK. Insulin secretory dysfunction and insulin resistance in the pathogenesis of Korean type 2 diabetes mellitus. Metabolism. 2001;50:590–3.
Matsumoto K, Miyake S, Yano M, Ueki Y, Yamaguchi Y, Akazawa S, Tominaga Y. Glucose tolerance, insulin secretion, and insulin sensitivity in nonobese and obese Japanese subjects. Diabetes Care. 1997;20:1562–8.
Suzuki H, Fukushima M, Usami M, Ikeda M, Taniguchi A, Nakai Y, et al. Factors responsible for development from normal glucose tolerance to isolated postchallenge hyperglycemia. Diabetes Care. 2003;26:1211–5.
Kashiwagi A, Kasuga M, Araki E, Oka Y, Hanafusa T, Ito H, Tominaga M, Oikawa S, Noda M, Kawamura T, Sanke T, Namba M, Hashiramoto M, Sasahara T, Nishio Y, Kuwa K, Ueki K, Takei I, Umemoto M, Murakami M, Yamakado M, Yatomi Y, Ohashi H; Committee on the Standardization of Diabetes Mellitus-Related Laboratory Testing of Japan Diabetes Society. International clinical harmonization of glycated hemoglobin in Japan: From Japan Diabetes Society to National Glycohemoglobin Standardization Program values. J Diabetes Invest. 2012;3:39–40.
Liang K, Zeger S. Longitudinal data analysis of continuous and discrete responses for pre-post designs. Sankhya Ind J Stat. 2000;62:134–48.
Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–705.
Pi-Sunyer FX. The effects of pharmacologic agents for type 2 diabetes mellitus on body weight. Postgrad Med. 2008;120:5–17.
Inzucchi SE. Oral antihyperglycemic therapy for type 2 diabetes: scientific review. JAMA. 2002;287:360–72.
Acknowledgments
The authors wish to thank Alan Meehan and Kathleen Newcomb (Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ, USA) for their assistance in writing and preparing this paper for submission and publication.
Conflict of interest
Authors Kadowaki, Tajima, and Odawara were advisory board members for this study. Authors Minamide, Kawashima, and Yanagida are employees of ONO Pharmaceutical Co., Ltd., Japan, and authors Okamoto and Arjona Ferreira are employees of MSD K.K. or Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ, USA. This study was sponsored by MSD K.K., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ, USA, the manufacturer of sitagliptin, and by Ono Pharmaceutical Co., Ltd., Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study is registered in ClinicalTrials.gov: NCT00854035, “A Phase III, Randomized, Placebo-Controlled, Double-Blind Clinical Trial and Subsequent Open-Label, Extension Clinical Trial to Study the Efficacy and Safety of Addition of MK-0431/ONO-5435 in Japanese Patients With Type 2 Diabetes Mellitus Who Have Inadequate Glycemic Control on Diet/Exercise Therapy and Insulin Monotherapy”, http://clinicaltrials.gov/ct2/show/NCT00854035.
Appendix: MK-0431/ONO-5435-15 P106 Primary Investigator list:
Appendix: MK-0431/ONO-5435-15 P106 Primary Investigator list:
Yoshio Kurihara (Kurihara Diabetic Care Clinic), Takuma Narita(Akita University Hospital), Kenji Yaginuma (Social Insurance Nihonmatsu Hospital), Tadasu Kasahara (Johsai Hospital), Yoshihiko Suzuki (HDC Atlas Clinic), Yukiko Onishi (The Institute For Adult Diseases, Asahi Life Foundation), Tadashi Okabe (Okabe Clinic), Keiko Tsunoda (Ekimaetsunoda Clinic), Masaaki Miyamoto (Nippon Medical School Hospital), Mako Tougo (Senpo Tokyo Takanawa Hospital), Tomio Inoue (Ageo Central General Hospital), Manabu Narimiya (Nishisaitamachuo National Hospital), Koichi Shinohara (Yashio Central General Hospital), Sane Ishikawa (Saitama Medical Center Jichi Medical University), Nobuichi Kuribayashi (Misaki Naika Clinic), Shuichi Watanabe (Kobari General Clinic), Yoh Miyashita (Toho University Sakura Medical Center), Makoto Ujihara (National Hospital Organization Yokohama Medical Center), Ikuro Matsuba (Matsuba Clinic), Takashi Iizuka (Asahi Internal Medicine Department Clinic), Junro Fuse (Kosugi Central Clinic), Mizuki Kaneshiro (Tsuruma Kaneshiro Diabetes Clinic), Fuyuki Minagawa (Minagawa Medical Clinic), Yoshiya Katsura (Tokyo Medical University Ibaraki Medical Center), Tadashi Iitsuka (Ibaraki Seinan Medical Center Hospital), Atsushi Ueda (Namegata District General Hospital), Hideo Takahashi (Minamiakatsuka Clinic), Shuichi Fukuda (Wakakusa Clinic), Katsunori Suzuki (Saiseikai Niigata Daini Hospital), Yoshiko Funase (Aizawa Hospital), Michio Nakagawa (Matsumoto Nakagawa Hospital), Mitsuo Imura (Yaizu City Hospital), Akira Yamauchi (Medical Care Law Person Corporation Rikeikai Suruga Clinic), Shigeki Endo (Endo Clinic), Hideki Okamoto (Meitetsu Hospital), Genichi Watanabe (Watanabe Clinic), Takanobu Toriyama (Nagoya Kyoritsu Clinic), Nobuo Takahashi (Takahashi Family Clinic), Makoto Hayashi (Matsunami General Hospital), Kazuyoshi Nishino (Jujo Rehabilitation Hospital), Shizuo Kajiyama (Kajiyama Clinic), Takaaki Yoshimasa (Yoshimasa Diabetes & Endocrine Clinic), Masako Deguchi (Japanese Red Cross Kyoto Daini Hospital), Kazuyuki Tobe (Toyama University Hospital), Rika Usuda (Toyama Prefectural Central Hospital), Yasuro Kumeda (Minamiosaka Hospital, Keigaku-Kai, Social Medical Corporation), Toshihiko Shiraiwa (Shiraiwa Medical Clinic), Shizuka Kaneko (Takatsuki Red Cross Hospital), Masanori Emoto (Osaka City University Hospital), Akira Ichibangase (Komatu Hospital, Kyojin-Kai Medical Corporation), Akihisa Imagawa (Osaka University), Takashi Ishihara (Kobe City Medical Center General Hospital), Kenta Hara (Kobe University Hospital), Jun-Ichiro Miyagawa (Hyogo College Of Medicine), Masahiro Emoto (Yamaguchi University Hospital), Masahiro Matsumoto (Kitakyushu Municipal Medical Center), Kiyohide Nunoi (St.Mary’s Hospital), Daisuke Goto (Kyushu Central Hospital Of The Mutual Aid Association Of Public School Teachers), Makoto Kunisaki (Kunisaki Makoto Clinic), Kentaro Yamada (Kurume University Hospital).
About this article
Cite this article
Kadowaki, T., Tajima, N., Odawara, M. et al. Efficacy and safety of sitagliptin add-on therapy in Japanese patients with type 2 diabetes on insulin monotherapy. Diabetol Int 4, 160–172 (2013). https://doi.org/10.1007/s13340-013-0109-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13340-013-0109-z