
Abstract
White spot syndrome virus (WSSV) is a pathogen that has emerged globally affecting shrimp populations. Comparison of WSSV genome have shown the virus to share a high genetic similarity except for a few variable genomic loci that has been employed as markers in molecular epidemiology studies for determining the origin, evolution and spread in different geographical regions. Molecular genotyping of WSSV are based on genomic deletions associated with ORF23/24 and ORF14/15 variable regions and the three variable number of tandem repeat regions, ORF75, ORF94 and ORF125. Studies show the prevalence of several genotypes for WSSV with particular genotypes being more prevalent than others in a given geographical area. Deletions associated with ORF23/24 and ORF14/15 variable regions have proven to be of evolutionary significance. Fitness and virulence studies on different genotypes of WSSV suggest that all the strains of WSSV are equally virulent, but the one with smaller genomic size is the fittest. Studies also have shown that mixed genotype infection of WSSV correlates with lower disease outbreaks. This review focuses on the genotyping studies that were undertaken in elucidating WSSV evolution and epidemiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
White spot syndrome virus (WSSV) is the most serious viral pathogen of farmed shrimp, often leading to mass mortalities resulting in diminished shrimp production and severe economic loss to farmers worldwide. The virus has a broad host range and is reported to infect a wide range of aquatic animals that includes marine and brackish water crustaceans, penaeids, crabs, freshwater prawns and crayfish, aquatic arthropods and planktons [14, 21, 30, 52]. WSSV can be transmitted by both horizontal and vertical mode [10] and therefore thought to be a major cause for the rapid emergence and extensive spread of the virus globally. Electron microscopic studies have shown the virus to have a rod shaped nucleocapsid with a cross-hatched appearance, surrounded by a trilaminar envelope and having a unique tail like appendage at one end [8, 29, 54]. WSSV has been classified as a sole member of the genus Whispovirus and family Nimaviridae [25, 43]. The virus has a large circular double-stranded DNA genome of ~300 kb size and is one of the largest genomes of animal viruses described so far [3, 40, 42, 55]. Since its discovery in Taiwan in 1992 [4], the virus has spread rapidly causing significant losses to shrimp production in almost all Asian countries, Middle East, North, Central and South America and other major shrimp farming countries of the world [9, 20, 35]. Recently outbreaks have also been reported in Mozambique showing that the virus has spread also to the African continent [http://www.oie.int/international-standard-setting/specialists-commissions-groups/aquatic-animal-commission-reports/]. In India, severe mortalities of cultured shrimp due to this virus were reported along the east coast during 1994–1995 [1, 17].
Initial attempts to genotype WSSV based on their morphology and protein profile [6, 50], restriction fragment length polymorphism patterns [22, 26, 29, 49], dinucleotide compositional analysis [37], suggested that except for small genetic differences, the various geographical isolates are similar and closely related to each other. However, the publication of complete genome sequences of WSSV isolates from, Taiwan (WSSV-TW) [40], China (WSSV-CN) [55] and Thailand (WSSV-TH) [42] has let computational analysis to be carried out on the sequences. Alignment of the published sequences has showed that although the three isolates shared an overall identity of >99 %, there existed differences within the genome which could be categorized as major variable genomic loci into (i) a genomic region prone to large deletions (~13 kb), also referred to as variable ORF23/24 region (ii) a variable genomic region (ORF14/15) prone to recombination (iii) a transposase encoded genomic region (iv) variable number of tandem repeats (VNTRs) within the coding regions of ORF75, ORF94 and ORF125 (v) single nucleotide mutations, including deletion, insertion or single nucleotide polymorphisms (SNPs) which have been suggested as genetic markers for the successful study of WSSV diversity [23]. Among the variable loci, the variations associated with ORF23/24 and ORF14/15, prone to deletion and recombination events, have been suggested to be useful in identifying evolutionary relationships of WSSV strains [23], (Fig. 1) while the VNTRs associated within the minisatellite loci have been suggested for the study of its epidemiology [23, 28, 32]. In recent years, the large number of studies based on morphology, molecular characterization, morphogenesis and pathogenesis of WSSV isolates carried out in various parts of the world have been reviewed [9, 30]. In the present article, we review the currently available information related to genotyping studies undertaken for understanding the epidemiological aspects, evolution and spread of WSSV.

Schematic representation of the variable loci with respect to WSSV-TH genome
WSSV Molecular Epidemiological Studies
Although a number of variable microsatellites, minisatellites and megasatellites have been reported as potential genetic markers for the study of WSSV epidemiology [23, 36], genotyping of WSSV has been mainly based on VNTR loci associated with the DNA minisatellites and the SNPs within the repeat units (RUs). A number of studies on WSSV epidemiology have used either singularly or in combination the VNTR loci encoded by ORF75, ORF94 and ORF125 (corresponding to the WSSV-TH genome, Fig. 1), as markers to characterize WSSV variants [6, 12, 18, 24, 32, 38]. The RU and their copy numbers pertaining to the published complete genome sequences is presented in Table 1. While ORF94 and ORF125 consist of perfect RUs of 54 and 69 bp respectively, ORF75 comprises of a compound RU of 45 bp interspersed by a 57 bp unit [36].
Among the three minisatellites, ORF located between the ribonucleotide reductase genes rr1 and rr2 of WSSV has been most commonly used in genotyping [6, 12, 27, 28, 32, 39, 41, 53]. Wongteerasupaya et al. [53] first demonstrated a practical method for differentiating WSSV genotypes based on the VNTRs associated with ORF94 from shrimp samples collected from WSSV outbreak ponds in Thailand between 2000 and 2002. They reported a wide variation in the tandem repeat units, ranging from 6 to 20 RUs, classifying the WSSV into 12 different genotypes. The most frequently encountered 54 bp RU recorded in their study was 8 with an alternating thymine or guanine (T or G) SNP at position 36 within the repeat unit. In a similar work, Dieu et al. [6] reported the WSSV Vietnam isolates to contain 7–17 RUs and a SNP to occur at the 48th position rather than the 36th position in the RU. Genotyping studies with Brazilian WSSV isolates showed that they differed for the number of 54 bp RU when compared to isolates from other countries in the Americas [27]. Further, the Brazilian WSSV isolates differed for the nucleotide pattern at position 48. In India, Musthaq et al. [28] were the first to carry out genotyping of WSSV targeting the ORF94 region for samples collected from outbreak ponds during 2002–2004. Of the 84 WSSV-infected shrimp samples analysed they reported 8 different repeat groups having 54 bp RUs ranging from 6 to 13 within ORF94 with 8 repeats being the most frequent. The SNP recorded in their study was T or G at position 48 of the RU. Pradeep et al. [32] in a similar study in India, reported 13 different repeat types for the ORF94 ranging from 2 to 16 RUs. Although 7 RUs was the most common among their samples analysed, most samples collected from outbreak ponds were associated with only 2 RUs. The SNP recorded by Pradeep et al. [32] for the 54 bp RU was at position 48 who attributed the differences seen for the SNP belonging either to the 36th or 48th position of the ORF94 RU to be due to the way in which the RU position was calculated as there existed a partial repeat of 12 bp at the end of the 54 bp repeat. A recent study carried out by Walker et al. [45] on the Indian WSSV showed the presence of 31 different genotypes of WSSV based on the analysis of ORF94 VNTR with individual shrimp samples harboring more than one WSSV genotype. These results indicate the prevalence of variable WSSV genotypes in a geographical region with some genotypes being more prevalent.
The repeats within ORF75 are reported to be more stable in comparison to ORF94 [6]. ORF75 comprises of a compound repeat of lengths 45 bp repeated several times interspersed by a 57 bp sequence. Pradeep et al. [32] in their study with 106 WSSV Indian isolates reported that PCR amplification of the ORF75 region yielded a product size ranging between 320 and 778 bp with majority yielding an amplicon of 525 bp. Further, sequencing of the smallest and the largest amplicon was reported to have a compound repeat of (45)3 57(45)2 and (45)2 57(45)5 57(45) 57(45)2 respectively and varied from the genotype of WSSV-TH, WSSV-TW and WSSV-CN. Similar work with 4 Brazilian isolates showed the number of repeats within ORF75 to be 10 and 11, while for other American isolates tested it ranged between 6 and 15 (Table 2). Further sequencing of the smallest RU of the Brazilian ORF75 fragment exhibited a repeat pattern of (45) 102(45)2102(45)2102(45)2 while the one containing 11 RUs was (45)102(45)3102(45)2102(45)2 [27].
ORF125 contains a tandem repeat of 69 bp. The WSSV-TH and WSSV-CN are identical in their genotypes and contain 6 RUs of 69 bp, while WSSV-TW has 8 RUs of the same. Dieu et al. [7] reported WSSV Vietnam isolates to show variation in the number of RUs in the ORF125 loci which ranged between 4 and 10. Similarly, 13 repeat types ranging from 2 to 14 RUs were documented for WSSV Indian isolates with 4 RUs being more frequent and having SNPs at 6 nucleotide positions [29, 37, 40, 43, 55, 56] in the first RU and three SNPs in the 4th RU at positions 50, 53 and 61 [32]. The number of repeats recorded for this locus among Brazilian isolates was 8–9 while for other American countries between 7 and 11 [27]. The variations recorded in the number of RUs in ORF75, ORF94 and ORF125 regions among different geographical WSSV isolates has been listed in Table 2. The number and order of RUs within the ORF75 and ORF125 loci has been suggested to be suitable markers in studying the regional spread of this virus [6]. However, it has also been suggested that while a combined analysis of all three variable loci would be useful in differentiating and characterizing specific WSSV strains for general epidemiological studies, the best marker, with maximum variation would be ORF94, followed by ORF125 and ORF75 [32].
Fitness and Virulence Differences in WSSV
The geographical isolates of WSSV identified so far are very similar in morphology and proteome [15, 29, 50]. Although direct comparisons have not been made, preliminary studies indicate that there seems to be little difference in virulence between various WSSV isolates [19, 51]. Further there is no difference in host range or tissue tropisms between the various WSSV isolates characterized so far [2, 11, 52]. Few studies based on genotypic variations have been undertaken to track the genetic structure of WSSV populations in order to infer the virulence pattern followed by this virus. Marks et al. [24] compared the virulence between two different WSSV isolates from Thailand, WSSV-TH-96-II containing the largest genome known till date (~312 kb) and WSSV-TH containing the smallest genome (∼293 kb) and found that the WSSV with the smallest genome to be more virulent than the largest. Reports based on ORF54 bp VNTR loci have suggested that the strains having <9 RUs to be more virulent. However, in all these studies WSSV samples from non-outbreak ponds were not analysed [12, 28, 53]. A recent study from Andhra Pradesh, India, showed predominance of two genotypes (TRS8 and TRS18) in outbreak ponds of which WSSV genotype TSR18 was more frequently associated with disease outbreaks [46]. A study carried out by Waikhom et al. [44] suggested that WSSV genotypes can vary upon passage in different host and that differential passaging can cause variation in host susceptibility and virulence. This observation was not in accordance to the bioassay study carried out by Pradeep et al. [31] wherein they compared the virulence between three different WSSV strains having RUs 3, 6 and 14 in the ORF94 region and reported that all strains of WSSV are equally virulent, but the one with smaller genomic size was the fittest, probably owing to the replication advantage. Their study also showed that there was no change in genotype upon passaging in different host.
VNTRs have also been used to demonstrate the prevalence of multiple WSSV genotypes in crabs, wild and farmed shrimp in the same pond where disease outbreaks have been recorded [45]. A recent study showed that WSSV mixed-genotype infections correlate with lower outbreak incidence and that disease outbreaks correlate with single-genotype infections [13]. However, elaborate studies carried out in small-holder black tiger shrimp (Penaeus monodon) farms in Andhra Pradesh, India, showed evidence that WSSV occurred simultaneously as a multitude of co-circulating genotypes in plankton, wild shrimp and crabs, which moved in waves through these plankton and wild crustacean population [45–47]. Once they gained entry into grow-out ponds, the shrimp got simultaneously exposed to several of these circulating different genotypes, resulting in infection which could progress as disease outbreaks. However, their study failed to show clear evidence of the implication of a single genotype to be principally responsible for disease outbreaks in ponds or correlate the prevalence of WSSV genotype associated with plankton, crabs or wild shrimp to be a potential source of infection in farmed shrimp. Passaging of WSSV in different hosts can alter the pathogenicity and the sequences of tandem repeat regions [44] and therefore approaches incorporating all the VNTR loci could be used to assess the relationship and correlate with the genotypic diversity and disease outbreaks.
WSSV Evolutionary Studies
Although WSSV has been reported in cultured shrimp worldwide since 1992 [4], questions regarding the origin and spread of the virus, its evolution both genetically and biologically over time remain unanswered and still a mystery [48]. WSSV epidemiology studies have faced design and execution problems as conventional methods used for studying WSSV distribution and epidemiology, based on farmer reports have met difficulties in screening ponds and monitoring disease outbreaks for WSSV [5]. Additionally the prevalence of this virus in different hosts [14, 21] has made it difficult to track the evolution of this virus. Molecular epidemiology methods with suitable genetic markers are a potential approach, wherein the genetic relatedness among isolates within and between different sources could help infer the virus origin and understanding of WSSV epidemiology and evolution. Marks et al. [23] analysed the three complete sequenced genome and suggested that the genomic deletions associated with ORF23/24 and ORF14/15 variable loci (Fig. 1) are prone to recombination and therefore of particular evolutionary significance as they could be used to study patterns of virus spread. Further, assessing the deletions of variable length present among WSSV in different geographical locations, different host species, within regions and associated host species could help assess and compare the routes of transmission in different farming systems. Dieu et al. [6] used the differences associated with ORF14/15 and ORF23/24 to characterize the WSSV Vietnam (WSSV-VN) isolates and observed that among the two deletion loci, ORF23/24 variable region to be a better marker for determining patterns of WSSV spread at an intermediate spatiotemporal scale. The study also suggested that WSSV-VN isolates and the WSSV-TH have a common lineage, which branched off from WSSV-TW and WSSV-CN early on and could have entered Vietnam by multiple introductions. Further, the authors proposed a model which suggested the spread of WSSV from either side of the Taiwan Strait into Vietnam based on the gradually increasing deletions in the ORF23/24 variable region.
Ramos-Pardes et al. [34] showed the ORF14/15 hypervariable regions of the WSSV isolated from Litopenaeus vannamei in northwest Mexico to be involved in recombination events and genetically similar to a strain from India and suggested that the mutations associated with this region could be an adaptation strategy to infect other host species. Marks et al. [24] characterized a WSSV isolate originating from Thailand in 1996 (TH-96-II) and suggested it to contain the largest genome (∼312 kb). Analysis of TH-96-II further suggests that probably this genome is an ancestral genome from which WSSV-TW and WSSV-VN could have originated [33], (Fig. 2). The genomic shrinkage due to deletions in these variable loci suggest a gradual loss from the largest genome recorded (WSSV-TH-96-II) to the smallest WSSV-TH. A recent study has shown that the Indian WSSV isolates have a 10,970 bp deletion in the ORF23/24 region relative to WSSV-TW and WSSV-TH-96-II while the difference between WSSV-TW and WSSV-TH-96-II was 13,210 bp [33], (Fig. 3). The transposase sequence or VP35 gene reported to code for a nucleocapsid a part of the 13 kb deletion region [23] was seen to be absent in all the Indian WSSV isolates [33]. Absence of transposase sequence is also recorded in WSSV-CN, WSSV-TH, Vietnam and Brazilian isolates [6, 23, 27]. Analysis of the ORF14/15 regions of Indian isolates also revealed the presence of three different strains of WSSV of which, two had novel sequences, which would have probably evolved by recombination (Fig. 2). The Indian strains are closely related to Thailand strains suggesting movement of a putative ancestor from Thailand to other parts of the world including India [33], (Fig. 4). Genotyping of Brazilian isolates showed WSSV to have a genomic deletion of 11,453 bp when compared to WSSV-TW isolate [27]. Similarly the Chinese WSSV isolates were reported to have deletions of 1,168, 5,657, 9,316 and 11,093 bp respectively in the variable region ORF23/24 in comparison to WSSV-TW and a deletion of 4,749 and 5,652 in the ORF14/15 region relative to TH-96-II [38, 39]. Studies so far on genetic characterization of WSSV isolates indicates a gradual loss in genomic size that has been progressively shrinking during its spread. The total size of deletions between the smallest (WSSV-TH) to the largest (WSSV-TH-96-II) being around 15 kbp, [7, 33]. Zwart et al. [56] in a recent study suggested that the rate of genome shrinkage decreases over time before attenuating and showed that WSSV spread did not follow a smooth pattern of geographic radiation, suggesting spread of WSSV over long distances to be due to commercial activities. Deletions in the variable region ORF23/24 have been suggested to play an important function in WSSV virulence [19]. Studies provide support for a link between virulence and WSSV fitness to be dependent on genome size, with smaller genomes exhibiting higher virulence and competitive fitness as compared to larger genomes [19, 24, 38]. It has been hypothesized that WSSV with smaller genomes are more stable and replicate more efficiently than those with larger genomes, probably responsible for increased fitness seen by which the virus gets easily established in shrimp aquaculture systems [24, 31, 39]. Considering the continuing worldwide emergence of WSSV it would be important to conduct molecular epidemiology investigation in all countries affected by WSSV to help elucidate the pattern of emergence and global spread of this virus.

Schematic representation of the ORF 14/15 variable region of WSSV strains from different geographical regions. Fragment lengths are indicated in each box and are as reported in the NCBI database. Deletions in the sequence are represented by dotted arrow (Ref. [33])

Schematic representation of the ORF 23/24 variable region of WSSV strains from different geographical regions. Lengths of the fragments are indicated above as boxes and deletions represented by dotted line. Nucleotide positions with respect to WSSV-TW genome are symbolized as Ф (Ref. [33])

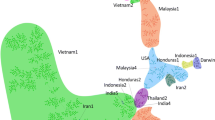
Map showing the possible geographical origin and transmission of WSSV in Southeast-Asia (Ref. [33]) Map source: www.nationsonline.org
Conclusion
WSSV has consistently been the most significant pathogen of cultivable shrimp and a major threat to shrimp farming industry across the globe. The sequencing of the three complete genome of WSSV from Thailand, Taiwan and China has paved way for the better understanding of WSSV epidemiology, movement and factors associated with virulence. Studies suggest that genotyping studies based on VNTR markers could be useful for the identification of potential sources of WSSV infection and tracking the patterns of disease while, deletions in the ORF14/15 and ORF23/24 variable regions can be used as markers to study patterns of virus spread over space and time. However, an integrated study on transmission, epidemiology and evolution of WSSV needs to be carried out, for better understanding and to develop strategies for prevention and management of this disease.
References
Anonymous. SEMBV—an emerging viral threat to cultured shrimp in Asia. CP Shrimp News. 1995;3:2–3.
Chen LL, Lo CF, Chiu YL, Chang CF, Kou GH. Natural and experimental infection of white spot syndrome virus (WSSV) in benthic larvae of mud crab Scylla serrata. Dis Aquat Org. 2000;40:157–61.
Chen LL, Wang HC, Huang CJ, Peng SE, Chen YG, Lin SJ, Chen WY, Dai CF, Yu HT, Wang CH, Lo CF, Kou GH. Transcriptional analysis of the DNA polymerase gene of shrimp white spot syndrome virus. J Virol. 2002;301:136–47.
Chou HY, Huang CY, Wang CH, Chiang HC, Lo CF. Pathogenicity of a baculovirus infection causing white spot syndrome in cultured penaeid shrimp in Taiwan. Dis Aquat Org. 1995;23:165–73.
Corsin F, Phi TT, Phuoc LH, Tinh NTN, Hao NV, Mohan CV, Turnbull JF, Morgan KL. Problems and solutions with the design and execution of an epidemiological study of white spot disease in black tiger shrimp (Penaeus monodon) in Vietnam. Prev Vet Med. 2002;53:117–32.
Dieu BTM, Marks H, Siebenga JJ, Goldbach RW, Zuidema D, Duong TP, Vlak JM. Molecular epidemiology of white spot syndrome virus within Vietnam. J Gen Virol. 2004;85:3607–18.
Dieu BTM, Marks H, Zwart MP, Vlak JM. Evaluation of white spot syndrome virus variable DNA loci as molecular markers of virus spread at intermediate spatiotemporal scales. J Gen Virol. 2010;91:1164–72.
Durand S, Lightner DV, Redman RM, Bonami JR. Ultrastructure and morphogenesis of white spot syndrome baculovirus (WSSV). Dis Aquat Org. 1997;29:205–11.
Escobedo-Bonilla CM, Alday-Sanz V, Wille M, Sorgeloos P, Pensaert MB, Nauwynck HJ. A review on the morphology, molecular characterization, morphogenesis and pathogenesis of white spot syndrome virus. J Fish Dis. 2008;31:1–18.
Flegel TW. Major viral disease of the black tiger prawn (Penaeus monodon) in Thailand. World J Microbiol Biotechnol. 1997;13:433–42.
Hameed AS, Balasubramanian G, Musthaq SS, Yoganandhan K. Experimental infection of twenty species of Indian marine crabs with white spot syndrome virus (WSSV). Dis Aquat Org. 2003;57:157–61.
Hoa TT, Hodgson RA, Oanh DT, Phuong NT, Preston NJ, Walker PJ. Genotypic variations in tandem repeat DNA segments between ribonucleotide reductase subunit genes of white spot syndrome virus (WSSV) isolates from Vietnam. Diseases in Asian Aquaculture V, Fish health section, Asian Fisheries Society, Manila. 2005;339–51.
Hoa TTT, Zwart MP, Phuong NT, Oanh DTH, Jong MCM, Vlak JM. Mixed-genotype white spot syndrome virus infections of shrimp are inversely correlated with disease outbreaks in ponds. J Gen Virol. 2011;92:675–80.
Hossain MS, Chakraborty A, Joseph B, Otta SK, Karunasagar I, Karunasagar I. Detection of new hosts for white spot syndrome virus of shrimp using nested polymerase chain reaction. Aquaculture. 2001;198:1–11.
Huang C, Zhang L, Zhang J, Xiao L, Wu Q, Chen D, Li JK. Purification and characterization of white spot syndrome virus (WSSV) produced in an alternate host: Crayfish, Cambarus clarkii. Virus Res. 2001;76:115–25.
John KR, George MR, Iyappan T, Thangarani AJ, Jeyaseelan MJP. Indian isolates of white spot syndrome virus exhibit variations in their pathogenicity and genomic tandem repeats. J Fish Dis. 2010;33:749–58.
Karunasagar I, Otta SK, Karunasagar I. Histopathological and bacteriological study of white spot syndrome of Penaeus monodon along the west coast of India. Aquaculture. 1997;153:9–13.
Kiatpathomchai W, Taweetungtragoon A, Jittivadhana K, Wongteerasupaya C, Boonsaeng V, Flegel TW. Target for standard Thai PCR assay identical in 12 white spot syndrome virus (WSSV) types that differ in DNA multiple repeat length. J Virol Methods. 2005;130:79–82.
Lan Y, Lu W, Xu X. Genomic instability of prawn white spot bacilliform virus (WSBV) and its association to virus virulence. Virus Res. 2002;90:269–74.
Lightner DV. Virus diseases of farmed shrimp in the Western Hemisphere (the Americas): a review. J Invertebr Pathol. 2011;106:110–30.
Lo CF, Ho CH, Peng SE, Chen CH, Hus HC, Chiu YL, Chang CF, Liu KF, Su MS, Wang CH, Kou GH. White spot syndrome baculovirus (WSBV) detected in cultured and captured shrimp, crab and other arthropods. Dis Aquat Org. 1996;27:215–25.
Lo CF, Hsu HC, Tsai MF, Ho CH, Peng SE, Kou GH, Lightner DV. Specific genomic DNA fragment analysis of different geographical clinical samples of shrimp white spot syndrome virus. Dis Aquat Org. 1999;35:175–85.
Marks H, Goldbach RW, Vlak JM, van Hulten MCW. Genetic variation among isolates of white spot syndrome virus. Arch Virol. 2004;149:673–97.
Marks H, van Duijse JJA, Zuidema D, van Hulten MCW, Vlak JM. Fitness and virulence of an ancestral white spot syndrome virus isolate from shrimp. Virus Res. 2005;110:9–20.
Mayo MA. A summary of taxonomic changes recently approved by ICTV. Arch Virol. 2002;147:1655–63.
Moon CH, Do JW, Cha SJ, Yoon WJ, Kim SB, Ko MS, Park MA, Kim JW, Sohn SK, Lee JH, Park JAW. Highly conserved sequences of three major virion proteins of a Korean isolate of white spot syndrome virus (WSSV). Dis Aquat Org. 2003;53:11–3.
Muller IC, Andrade TP, Tang-Nelson KF, Marques MR, Lightner DV. Genotyping of white spot syndrome virus (WSSV) geographical isolates from Brazil and comparison to other isolates from the Americas. Dis Aquat Organ. 2010;88:91–8.
Musthaq SS, Sudhakaran R, Ahmed VP, Balasubramanian G, Hameed AS. Variability in the tandem repetitive DNA sequence of white spot syndrome virus (WSSV) genome and stability of VP28 gene to detect different isolates of WSSV from India. Aquaculture. 2006;256:34–41.
Nadala ECB Jr, Loh PC. A comparative study of three different isolates of white spot virus. Dis Aquat Org. 1998;33:231–4.
Paz SA. White spot syndrome virus: an overview on an emergent concern. Vet Res. 2010;41:43.
Pradeep B, Karunasagar I, Karunasagar I. Fitness and virulence of different strains of white spot syndrome virus. J Fish Dis. 2009;32:801–5.
Pradeep B, Shekar M, Gudkovs N, Karunasagar I, Karunasagar I. Genotyping of white spot syndrome virus prevalent in shrimp farms of India. Dis Aquat Org. 2008;78:189–98.
Pradeep B, Shekar M, Karunasagar I, Karunasagar I. Characterization of variable genomic regions of Indian white spot syndrome virus. Virology. 2008;376:24–30.
Ramos-Paredes J, Grijalva-Chon JM, de la Rosa-Vélez J, Enríquez-Paredes LM. New genetic recombination in hypervariable regions of the white spot syndrome virus isolated from Litopenaeus vannamei (Boone) in northwest Mexico. Aqua Res. 2011;42:1–10.
Rodriguez J, Bayot B, Amano Y, Panchana F, de Blas I, Alday V, Calderon J. White spot syndrome virus infection in cultured Penaeus vannamei (Boone) in Ecuador with emphasis on histopathology and ultrastructure. J Fish Dis. 2003;26:439–50.
Shekar M, Karunasagar I, Karunasagar I. A computer-based identification of variable number tandem repeats in white spot syndrome virus genomes. Curr Sci. 2005;89:882–7.
Shekar M, Karunasagar I, Karunasagar I. Abundance, composition and distribution of simple sequence repeats and dinucleotide compositional bias within WSSV genomes. J Genet. 2007;86:69–73.
Tan Y, Xing Y, Zhang H, Feng Y, Zhou Y, Shi Z. Molecular detection of three shrimp viruses and genetic variation of white spot syndrome virus in Hainan Province, China, in 2007. J Fish Dis. 2009;32:777–84.
Tan Y, Shi Z. Genotyping of white spot syndrome virus in Chinese cultured shrimp during 1998–1999. Virologica Sin. 2011;26:123–30.
Tsai MF, Lo CF, van Hulten MC, Tzeng HF, Chou CM, Huang CJ, Wang CH, Lin JY, Vlak JM, Kou GH. Transcriptional analysis of the ribonucleotide reductase genes of shrimp white spot syndrome virus. Virology. 2000;277:92–9.
van Hulten MC, Tsai MF, Schipper CA, Lo AF, Kou GH, Vlak JM. Analysis of a genomic segment of white spot syndrome virus of shrimp containing ribonucleotide reductase genes and repeat regions. J Gen Virol. 2000;81:307–16.
van Hulten MC, Witteveldt J, Peters S, Kloosterboer N, Tarchini R, Fiers M, Sandbrink H, Lankhorst RK, Vlak JM. The white spot syndrome virus DNA genome sequence. Virology. 2001;286:7–22.
Vlak JM, Bonami JR, Flegel TW, Kou GH, Lightner DV, Lo CF, Loh PC, Walker PW. Nimaviridae. In: Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA, editors. Virus taxonomy. Eighth Report of the International Committee on taxonomy of viruses. Amsterdam: Elsevier; 2005. p. 187–92.
Waikhom G, John KR, George MR, Jeyaseelan MJP. Differential host passaging alters pathogenicity and induces genomic variation in white spot syndrome virus. Aquaculture. 2006;261:54–63.
Walker PJ, Gudkovs N, Pradeep B, Raj VS, Sergeant E, Chandra Mohan AB, Ravibabu G, Umesh NR, Karunasagar I, Santiago TC, Mohan CV. Longitudinal disease studies in small-holder black tiger shrimp (Penaeus monodon) farms in Andhra Pradesh, India. III. A complex dynamic of WSSV infection and WSSV genotype distribution in farmed shrimp and wild crustaceans. Aquaculture. 2011;319:319–27.
Walker PJ, Gudkovs N, Mohan CV, Raj VS, Pradeep B, Sergeant E, Chandra Mohan AB, Ravibabu G, Karunasagur I, Santiago TC. Longitudinal disease studies in small-holder black tiger shrimp (Penaeus monodon) farms in Andhra Pradesh, India. II. Multiple WSSV genotypes associated with disease outbreaks in ponds seeded with uninfected postlarvae. Aquaculture. 2011;319:18–24.
Walker PJ, Gudkovs N, Padiyar PA, Raj VS, Pradeep B, Sergeant E, Chandra Mohan AB, Ravibabu G, Vijayan KK, Karunasagar I, Santiago TC, Mohan CV. Longitudinal disease studies in small-holder black tiger shrimp (Penaeus monodon) farms in Andhra Pradesh, India. I. Correlation of WSSV infection with risk of crop failure. Aquaculture. 2011;318:277–82.
Walker PJ, Winton JR. Emerging viral diseases of fish and shrimp. Vet Res. 2010;41:51.
Wang Q, Nunan LM, Lightner DV. Identification of genomic variations among geographic isolates of white spot syndrome virus using restriction analysis and southern blot hybridization. Dis Aquat Org. 2000;43:175–81.
Wang Q, Poulos BT, Lightner DV. Protein analysis of geographic isolates of shrimp white spot syndrome virus. Arch Virol. 2000;145:263–74.
Wang Q, White BL, Redman RM, Lightner DV. Per os challenge of Litopenaeus vannamei postlarvae and Farfantepenaeus duorarum juveniles with six geographic isolates of white spot syndrome virus. Aquaculture. 1999;170:179–94.
Wang YC, Lo CF, Chang PS, Kou GH. Experimental infection of white spot baculovirus in some cultured and wild decapods in Taiwan. Aquaculture. 1998;164:221–31.
Wongteerasupaya C, Pungchai P, Withyachumnarnkul B, Boomsaeng V, Panyim S, Flegel TW, Walker PJ. High variation in repetitive DNA fragment length for white spot syndrome virus (WSSV) isolates in Thailand. Dis Aquat Org. 2003;54:253–7.
Wongteerasupaya C, Vickers JE, Sriurairatana S, Nash GL, Akarajamorn A, Boonsaeng V, Panyim S, Tassanakajon A, Withyachumnarnkul B, Flegel TW. A non-occluded, systemic baculovirus that occurs in cells of ectodermal and mesodermal origin and causes high mortality in the black tiger prawn Penaeus monodon. Dis Aquat Org. 1995;21:69–77.
Yang F, He J, Lin XH, Li Q, Pan D, Zhang XB, Xu X. Complete genome sequence of the shrimp white spot bacilliform virus. J Virol. 2001;75:11811–20.
Zwart MP, Dieu BTM, Hemerik L, Vlak JM. Evolutionary trajectory of white spot syndrome virus (WSSV) genome shrinkage during spread in Asia. PLoS ONE. 2010;5:13400–11.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shekar, M., Pradeep, B. & Karunasagar, I. White spot syndrome virus: Genotypes, Epidemiology and Evolutionary Studies. Indian J. Virol. 23, 175–183 (2012). https://doi.org/10.1007/s13337-012-0078-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-012-0078-z